Neuromuskulárne ochorenia. Svalové ochorenia - ochorenia nervového systému u detí.
Zápal je veľmi bolestivé ochorenie, ktoré je charakterizované zápalovými procesmi v kostrovom svalstve, s rôznymi príčinami. V lekárskej praxi sa to nazýva myozitída. Hlavnou charakteristikou je všeobecná svalová slabosť, ktorá je vyvolaná zápalovými procesmi vo svalových tkanivách zodpovedných za motorickú záťaž. Tento zápal zahŕňa ďalšie typy spojené s infekciami, toxínmi a často s škodlivé účinky lieky. Hlavné zápalové procesy sú viacnásobné a spojené s povrchovou léziou.
Kto má väčšiu pravdepodobnosť, že dostane myozitídu?
Podľa dlhodobých pozorovaní sa predpokladá, že výskyt takéhoto ochorenia je od dvoch do desiatich prípadov na jeden milión priemernej ľudskej populácie. Ochorenie je charakterizované dvoma vekovými vrcholmi výskytu. Prvý nastáva medzi piatym a šestnástym rokom života. Druhý vrchol pripadá na viac seniorská skupinaštyridsať až šesťdesiat rokov. Odhalila sa aj rodová priorita v chorobe, na túto chorobu sú viac choré ženy, pomer chorých žien k chorým mužom sa pohybuje v priemere od dvoch do troch u žien po jednu u mužov.
Aké sú príčiny zápalu?
Až doteraz neboli presné príčiny tohto ochorenia objasnené. Ale práve sezónny vplyv z hľadiska frekvencie výskytu priamo nenaznačuje nárast počtu ochorení v zimnom období alebo skoro na jar, a to je práve obdobie rozkvetu prekračovania epidemických prahov infekčných ochorení. Ak vezmeme do úvahy genetický vplyv na výskyt týchto zápalov, potom sa vyskytujú najmä u jednovaječných dvojčiat a príbuzných v jednej línii, ktoré už takéto ochorenie mali. Takéto prenášanie génovej informácie zvyčajne nie je spojené so samotnou chorobou, ale je spojené so špecifickými poruchami imunitného systému.
Prvé príznaky myozitídy
Hlavnými ukazovateľmi priebehu ochorenia sú poruchy imunitných reakcií na bunkovej úrovni. Imunopatologické vyšetrenie zapálenej oblasti svalov odhaľuje patologickú infiltráciu niektorých bunkových tiel, ktoré sú v počiatočnom aktívnom stave, a uvoľňuje toxín, ktorý ovplyvňuje procesy obnovy. S kožným prejavom ochorenia je možné identifikovať určité patogény, ktoré porušujú celkovú bunkovú rezistenciu. Zisťuje sa aj protilátková reakcia, ktorá má v niektorých prípadoch negatívne reakcie na cievy.
V počiatočnom priebehu ochorenia sú časté návštevy pacientov pre malátnosť a veľkú slabosť a zápalové procesy na koži. V priebehu niekoľkých týždňov dochádza k nárastu počiatočných príznakov a nárastu slabosti a, motorické svaly ach ruky a nohy. V prípade ochorenia mladého organizmu sa pozorujú akútne počiatočné príznaky sprevádzané výraznými zmenami v konštitúcii tela, buď úbytkom hmotnosti, alebo febrilnými javmi.
Ak vezmeme do úvahy starších pacientov, potom pozorujú postupný nárast slabosti svalov, niekedy v priebehu niekoľkých rokov. Menej často sa prejavujú lézie povrchovej kože. U niektorých pacientov sa môžu objaviť aj špecifické znaky, ktoré sú sprevádzané kŕčmi krvných ciev a poruchami krvného obehu, poruchami kostného tkaniva a dýchacími ťažkosťami, ku ktorým dochádza v dôsledku niektorých zápalových procesov v pľúcach.
Ako sa choroba prejavuje?
Z klinického hľadiska je prvým prejavom tohto ochorenia celková slabosť všetkých svalových skupín zodpovedných za motorickú činnosť a hornej a dolnej skupiny týchto svalov, ako aj krčnej oblasti. Tieto poruchy vedú k tomu, že pacient sa v počiatočnej fáze po dlhom odpočinku veľmi ťažko hýbe, ťažko vstáva zo sedu, nastupuje alebo vystupuje z transportu alebo pri iných denných pohyboch. Zboku sú viditeľné poruchy chôdze, stáva sa nejasným, málo koordinovaným.
Sú prípady, keď sa pacient nedokáže sám dostať z postele. Svalový zápal môže postihnúť aj oblasť spojenú s prehĺtaním a hlasom, môže to byť sprevádzané akútnymi ťažkosťami pri prehĺtaní, ktoré končí výbušným kašľom. U niektorých pacientov sa tiež vyvinie dermatomyozitída, ktorá má prejavy vo forme oblastí so zvýšenými vyrážkami na určitých častiach tela alebo na tvári alebo krku a na iných častiach tela. Existujú aj iné kožné prejavy, ktoré priamo poukazujú na priebeh ochorenia, môžu sa kombinovať aj s prejavmi polymyozitídy, môžu to byť šupinaté a začervenané miesta na koži alebo dokonca sprevádzané kožnými mikrotrhlinkami na niektorých miestach horných končatín.
Ukladanie vápenatých solí v mäkkých tkanivách
Ukladanie solí v mäkkých tkanivách (kalcifikácia) je zvyčajne produktom dlhodobého ochorenia, v neskorších štádiách zápalových procesov. Formácie sa objavujú a sú zvyčajne lokalizované pod kožou alebo v miestach spájania svalov, nad kĺbovými časťami kolien a lakťov, nad falangami prstov a mnohými ďalšími a dokonca aj v niektorých oblastiach zadku.
Zápalové procesy v pľúcach s myozitídou
Jasným počiatočným znakom zápalových procesov v oblasti pľúc s myozitídou je dýchavičnosť, zvyčajne je spojená s porušením svalov bránice alebo abnormálnou srdcovou funkciou alebo so zápalom spojeným s toxínmi obsiahnutými v liekoch, ktoré pacient užíva. . Zápalové procesy sú tiež sprevádzané kašľom alebo nedostatočnou respiračnou aktivitou. V obzvlášť ťažkých prípadoch sa môže vyvinúť dusivý zápal pľúc.
Ako myozitída ovplyvňuje srdce?
Zápalové procesy vyskytujúce sa v srdcovom svale s myozitídou zvyčajne prebiehajú bez výrazných symptómov. Iba pri špeciálnej štúdii s použitím kardiogramu sa pozorujú niektoré zlyhania rytmov rytmu, ktoré sa zvyčajne pripisujú oblastiam iných ochorení. Srdcové zlyhanie sa prakticky nepozoruje.
Diagnóza zápalu v laboratórnych štúdiách
Pri vykonávaní krvných testov nie sú pozorované závažné zmeny. Ďalšie analýzy odhaľujú odchýlky, ktoré sú charakteristické pre poruchy svalov zodpovedných za motorickú aktivitu (CPK). V niektorých priebehoch ochorenia sa pozorujú takmer u všetkých pacientov. Sýtosť tohto indikátora sa môže zvyšovať, kým sa počas klinického vyšetrenia nezistia jasné počiatočné príznaky. Ale niekedy môže byť tento indikátor v normálnom rozmedzí a dokonca ani nenaznačuje závažný zápal svalového tkaniva. Existuje aj indikátor s nízkou motorickou schopnosťou, ktorý naznačuje niektoré možné bežné príznaky hepatitídy (transaminázy).
Liečba zápalu svalov
odhalené v praxi. Že zápal svalov je choroba, ktorá prebieha podľa periodickej schémy, teda s exacerbáciami. Bolesti vznikajúce pri iných chronických ochoreniach sú konštantné a v prípade tohto ochorenia sú periodické a neustále neobťažujú. Práve myozitída má dve štádiá priebehu ochorenia - chronické a akútne. Pri správnej liečbe akútne štádium ochorenia plynule prechádza do chronického, nie až tak bolestivého. V chronickom štádiu ochorenia sa zápalové procesy stávajú citlivými na sezónne zmeny alebo klimatické zmeny. Chronický zápal môže byť spôsobený nielen ako štádium po akútnej fáze myozitídy. Ale aj s niektorými chorobami infekčného charakteru. Preto si ľudia často ani neuvedomujú myozitídu, keď sú chorí s obvyklou infekčnou chrípkou, ale po zotavení sa objavia známky poškodenia svalov. Akútny zápal svalov sa môže prejaviť v lokálnej lokalizácii.
Je veľmi ťažké odhaliť chorobu myozitídu, pretože je prísne periodická. Ale pri prvých príznakoch musíte kontaktovať špecialistu. Malo by sa to robiť z niekoľkých dôvodov, ale prvým by mala byť definícia presnej diagnózy a potom určenie optimálnej liečby, hlavná vec je, že je systémová.
Liečba zápalu svalov môže niesť správne rozloženie záťaže na organizmus s fyzickým dopadom na ochorenie. V niektorých prípadoch sa používajú športové záťaže, ktoré sa striedajú s obdobiami úplnej relaxácie. Prvoradé pri liečbe myozitídy je zistiť príčiny zápalu. Na základe výskumu medikamentózna liečba pomocou špecializovaných liekov. Zvyčajne je na liečbu predpísaný komplex liekov, ale v každom systéme sú zahrnuté lieky proti bolesti na báze analgetík a liekov, ktoré zabraňujú zápalovým procesom. Používa sa veľa liekov, ale hlavné sú Diclofenac, Ketonal, Nurofen.
S lokálnymi alebo lokálnymi ohniskami ochorenia sa používajú špeciálne prípravky, ktoré sú predpísané vo forme masti otepľovacieho typu: Apizatron, Nikoflex, Finalgon. Pri liečbe zápalových procesov u detí sa zvyčajne predpisujú špecializované prípravky so zníženou dávkou liečivých zložiek, v tomto prípade sú dobré recenzie o lieku Doctor MOM.
V obdobiach, keď dochádza k exacerbácii priebehu ochorenia, v žiadnom prípade by ste nemali zaťažovať telo fyzickou námahou a vo všeobecnosti musíte dodržiavať odpočinok v posteli. Toto má prvoradý význam pri zápalových procesoch v oblasti miechových motorických svalov. Pri takejto exacerbácii sú potrebné nasledujúce analgetické lieky a lieky, ktoré zabraňujú zápalovým procesom, zoznam takýchto liekov zahŕňa: Brufen, Reopirin, Indometacin.
Takáto liečba môže byť účinná iba v takom prípade. Ak je systémový a používa sa v kombinácii s inými účinnými metódami a prostriedkami. Z toho vyplýva, že popri užívaní liekov je potrebné vykonávať aj fyzioterapeutické procedúry a je veľmi žiaduce, aby sa využívali cvičenia zahrnuté v liečebno-preventívnej telesnej výchove. Malo by sa však pamätať na to, že pri akútnej myozitíde motorických svalov chrbtice je takáto telesná výchova prísne kontraindikovaná. Pri zvýšení celkovej telesnej teploty v priebehu ochorenia je potrebné užívať lieky, ktoré ju znižujú a dbať na obmedzenie kontaktu s chladnou atmosférou.
V prípadoch, keď sú zápalové procesy hornej časti svalov zodpovedné za motorickú záťaž na krku spôsobené septickými léziami, potom po definitívnom výkone je potrebné kontaktovať praktizujúceho chirurga, ktorý je povinný túto zónu otvoriť v r. operabilným spôsobom a odstrániť celú infekciu. Taktiež pri tomto type zápalu svalov je akýkoľvek druh masáže prísne zakázaný.
Každý typ myozitídy má svoje vlastné vlastnosti, ktoré sú charakteristické len pre neho. Napríklad zápalový proces krčnej oblasti svalovej skupiny sa lieči bez zvláštnych komplikácií a je relatívne jednoduchý, ak sa s touto liečbou nezačalo pri pokročilom ochorení, ale hneď po objavení sa prvých príznakov. Pri liečbe tohto typu myozitídy odborníci zvyčajne predpisujú sedavú liečbu alebo najlepšie pokoj na lôžku. Ošetrujúci špecialisti predpisujú komplex liekov vrátane otepľovacích mastí, ktoré sa vtierajú do oblasti zápalu a užívajú lieky, ktoré zabraňujú zápalovým procesom.
V praxi dáva vynikajúci výsledok takzvaná "novokaínová blokáda". Proces tejto udalosti spočíva v bodovej aplikácii anestetika okolo zapáleného miesta s Novocainom a špeciálnym hormónom. To však dáva pozitívny účinok iba v prípadoch, keď pacient nemá žiadne alergické reakcie a kontraindikácie. Pre relaxačnú procedúru existuje aj metóda naťahovania svalových a väzivových tkanív. Ide o úplne novú metódu, ktorá dáva vynikajúce výsledky a už bola testovaná v mnohých lekárskych ústavoch.
V priloženom videu môžete odhaliť, prečo sa šľachy zapália.
Ale najjednoduchšie odporúčania, ktoré dobrý odborník sprevádza každého potenciálneho pacienta, sú odporúčania chrániť kritické oblasti svalov pred chladom, nestagnovať a nezostávať dlho v jednej statickej polohe v ľahu aj v sede. Vyberte si iba pohodlné pózy, v ktorých svaly netrpia, vylúčte prievan a vykonajte všeobecné posilňovacie cvičenia. Tieto jednoduché a pomerne nekomplikované odporúčania vám pomôžu zbytočne sa nevystavovať zápalovým procesom a myozitíde.
Neuromuskulárne ochorenia sú charakterizované poruchou funkcie vôľových svalov, stratou alebo znížením motorickej kontroly, čo môže nastať v dôsledku poškodenia samotných svalov a môže byť sekundárne – v dôsledku dysfunkcie nervovosvalového spojenia, poškodenia periférnych nervov alebo motorických neurónov . miecha. V klinickom obraze niektorých nervovosvalových ochorení môžu byť príznaky poškodenia motorických jadier mozgového kmeňa. Lézie v iných oblastiach nervový systém, ktoré vedú k porušeniu motorickej kontroly, najmä pyramídového traktu, podľa všeobecne uznávanej definície nepatria medzi nervovosvalové ochorenia.
Najčastejšími príznakmi nervovosvalových ochorení sú slabosť, znížený objem svalov (atrofia), mimovoľné svalové zášklby, kŕče, necitlivosť, mravčenie atď. Dysfunkcia nervovosvalového spojenia môže spôsobiť poklesnuté viečka (ptózu), dvojité videnie (diplopiu) a iné príznaky svalovej slabosti, ktoré sa zvyšujú počas dňa. Pri niektorých chorobách môže byť narušené prehĺtanie a dokonca aj dýchanie.
Svalové ochorenia: Symptómy
- progresívne svalové dystrofie je genetické dedičné svalové ochorenie, ktorého príznaky sa zvyčajne objavujú v dojčenskom veku resp detstvo menej často u dospelých. Svalová slabosť sa postupne zvyšuje, najmä v ľubovoľných svaloch. Do tejto skupiny patrí Beckerova svalová dystrofia, vrodená svalová dystrofia, distálna svalová dystrofia, Duchennova svalová dystrofia (najčastejšia forma svalovej dystrofie u detí), Emery-Dreyfusova svalová dystrofia, ramenno-skapulárna svalová dystrofia, myotonická svalová dystrofia (najčastejšia forma svalová dystrofia u dospelých), okulofaryngeálna myodystrofia.
- Zápalové myopatie- nazývaná aj myozitída, je založená na zápalovom procese, ktorý vedie k svalovej slabosti, pri ich vzniku sa zdôrazňuje úloha autoimunitných porúch, niekedy kombinovaných s inými autoimunitnými ochoreniami. Patria sem dermatomyozitída, polymyozitída, myozitída s inklúziami.
- Mitochondriálne myopatie- vznikajú v dôsledku štrukturálnych alebo biochemických defektov v mitochondriách. Kearnsov-Syreov syndróm, myoklonová epilepsia s „roztrhanými červenými vláknami“, mitochondriálna encefalomyopatia.
- Myotónia - vrodená myotónia alebo Thomsenova choroba, dystrofická myotónia, vrodená paramyotónia, neuromyotónia alebo Isaacsova choroba
- Iné myopatie – centrálne ochorenie srdca, myotubulárna myopatia, nemalinová myopatia, hyperkalemická a hypokaliemická periodická paralýza, endokrinné myopatie
Choroby nervovosvalového spojenia
Spôsobujú dysfunkciu normálneho synaptického prenosu vzruchov z nervových zakončení do svalových vlákien. Ochorenie môže byť založené na autoimunitnom procese.
- myasthenia gravis
- Lambertov-Eatonov syndróm
- vrodený myastenický syndróm
Ochorenia periférnych nervov
- Mononeuropatie - poškodenie jedného nervu, najčastejšou príčinou je kompresný efekt (tunelové syndrómy), traumatické poranenia
- Mnohopočetné mononeuropatie - multifokálne lézie periférnych nervov, zvyčajne spojené so systémovými alebo infekčnými ochoreniami, paraneoplastické syndrómy
- Polyneuropatie sú difúzne, symetrické lézie periférnych nervov, zvyčajne dominujú v distálnych častiach, takmer vždy sú v klinickom obraze poruchy citlivosti. Môžu byť akútne (Guillain-Barrého syndróm), chronické (chronická zápalová demyelinizačná polyneuropatia), získané (toxická, diabetická, paraneoplastická) alebo dedičné (peroneálna svalová atrofia alebo Charcot-Marie-Tousova choroba, Dejerine-Sottova choroba, Friedreichova ataxia).
- Plexopatie - lézie plexusov horných a dolných končatín (rameno a lumbosakrálne), ktorých najčastejšou príčinou sú traumatické alebo kompresné účinky
- Radikulopatia a polyradikulopatia - lézie motorických alebo senzorických miechových koreňov
Choroby motorický neurón
Progresívna degeneratívna lézia motorických neurónov, ktorá má najvýraznejšie za následok zhoršenú motorickú kontrolu horných alebo dolných končatín, ako aj bulbárne poruchy. Nástup najčastejšie v strednom veku, príznaky môžu zahŕňať slabosť končatín, ťažkosti s prehĺtaním, rečou, chôdzou, slabosťou tvárových svalov a svalové kŕče. Do tejto skupiny patria najmä:
- amyotrofická laterálna skleróza (ALS)
- spinálna svalová atrofia dospelých
- spinálna svalová atrofia u dojčiat alebo Werdnig-Hoffmannova choroba
- juvenilná spinálna svalová atrofia alebo Kugelberg-Welanderova choroba
- bulbospinálna svalová atrofia alebo Kennedyho choroba
Diagnóza vychádza z anamnézy ochorenia, dôkladné neurologické vyšetrenie, vo väčšine prípadov sa používa elektromyografická (EMG) štúdia, pri kombinácii s léziou centrálneho motorického neurónu alebo na jej vylúčenie možno použiť transkraniálnu magnetickú stimuláciu, ak je dedičná existuje podozrenie na formy, robí sa analýza DNA, autoimunitný charakter procesu vyžaduje stanovenie špecifických protilátok, možno vykonať biopsiu svalovej oblasti, pri primárnych svalových léziách sa kontroluje hladina kreatínfosfokinázy (CPK) a ultrazvukové vyšetrenie svaly a periférne nervy si v poslednej dobe získavajú na popularite. Diagnostický algoritmus, výber ďalších štúdií závisí od charakteristík klinického vzoru a lokalizácie lézie - sval, nerv, plexus, korene, motorické neuróny.
DEDIČNÉ CHOROBY NERVOVÉHO SYSTÉMU
PREDNÁŠKA 16
Degeneratívne ochorenia s prevládajúcou léziou nervovosvalového aparátu tvoria najväčšiu skupinu všetkých dedičných ochorení.
Mimoriadne dôležité a často rozhodujúce v diagnostike nervovosvalových ochorení sú výsledky elektrofyziologických a biochemických štúdií. Rovnako veľký je význam patomorfologických nálezov. Štúdium svalovej biopsie v svetelný mikroskop pomáha odlíšiť myogénnu atrofiu od neurogénnej atrofie. Histochemické vyšetrenie je nevyhnutné na odhalenie metabolických svalových lézií a elektrónová mikroskopia otvorila celú veľkú triedu ochorení – neprogresívnych myopatií.
Progresívne svalové dystrofie. Pojem svalové dystrofie je skupina geneticky podmienených porúch charakterizovaných progresívnymi degeneratívnymi zmenami svalových vlákien bez primárnej patológie periférneho (dolného) motorického neurónu.
Rôzne formy sa navzájom líšia typmi dedičnosti, načasovaním nástupu procesu, povahou a rýchlosťou jeho priebehu, originalitou topografie svalovej atrofie, prítomnosťou alebo absenciou pseudohypertrofie a retrakcií šliach a inými znakmi.
Väčšina svalových dystrofií je dobre klinicky študovaná Detailný popis vyrobený koncom minulého storočia. Ale napriek takmer storočnej histórii štúdia myodystrofie otázky ich patogenézy a liečby zostávajú dodnes nevyriešené. Veľké nádeje sa vkladajú do molekulárnej genetiky, pomocou ktorej sa už podarilo určiť umiestnenie génov mnohých nosologických foriem.
Diagnóza svalových dystrofií často predstavuje veľké ťažkosti. Existuje veľká variabilita klinických prejavov a malý počet členov rodiny sťažuje určenie typu dedičnosti.
Charakteristickým motorickým defektom u pacientov so svalovými dystrofiami je „kačacia“ chôdza: pacient kráča kolísavo na jednu stranu. Súvisí najmä so slabosťou sedacích svalov, predovšetkým stredných a malých, ktoré fixujú panvu voči stehennej kosti. Následkom ochorenia dochádza k záklonu panvy k nepodporujúcej nohe (Trendelenburgov fenomén) a kompenzačnému záklonu tela v r. opačná strana(Duchenneov fenomén). Pri chôdzi sa strana svahu neustále mení. Tieto zmeny je možné skontrolovať aj v Trendelenburgovom teste tak, že pacienta požiadate, aby zdvihol jednu nohu a pokrčil ju v pravom uhle v kolenných a bedrových kĺboch: panva na strane zdvihnutej nohy klesá (a nedvíha sa ako normálne) v dôsledku slabosti svalu gluteus medius opornej nohy.
Pacient s ťažkou svalovou slabosťou proximálnych svalov sa zdvihne z vodorovnej polohy a takmer sa neprevráti na brucho, potom, keď si položí ruky na podlahu, postaví sa na všetky štyri a potom si položí ruky na holene a potom na boky. , postupne sa narovnáva. Tento fenomén „vyberať si na vlastnú päsť“ sa nazýva Gowersov manéver. Často je spojená so slabosťou svalov gluteus maximus.
Duchennova myodystrofia. Pseudohypertrofická Duchennova svalová dystrofia sa vyskytuje častejšie ako všetky ostatné ochorenia svalového systému (30 na 100 000 živonarodených detí). Vyznačuje sa skorým nástupom a malígnym priebehom. Klasický obraz sa prejavuje zmenou chôdze u dieťaťa vo veku 2-5 rokov, vo veku 8-10 rokov už deti chodia s ťažkosťami, do 14-15 rokov sú väčšinou úplne imobilizované. U detí v nižšom veku sa prvotné príznaky prejavujú zaostávaním motorického vývinu: neskôr začínajú chodiť, nevedia behať a skákať. Pacienti zomierajú v 2. alebo 3. dekáde života.
Jedným z prvých príznakov ochorenia je zhutnenie lýtkových svalov a postupné zväčšovanie ich objemu v dôsledku pseudohypertrofie. Atrofia svalov stehna, panvového pletenca je často maskovaná dobre vyvinutým podkožným tukovým tkanivom. Postupne sa proces uberá smerom nahor a šíri sa za ramenný pletenec, chrbtové svaly a potom do proximálnych častí rúk.
V terminálnom štádiu sa svalová slabosť môže rozšíriť na svaly tváre, hltana a dýchacích svalov.
V pokročilom štádiu ochorenia existujú také charakteristické príznaky ako "kačacia chôdza"; zvýraznená bedrová lordóza, pterygoidné lopatky, symptóm „uvoľneného ramenného pletenca“. Typické sú skoré svalové kontraktúry a retrakcie šliach, najmä Achillových šliach. Kolenné reflexy vypadnú skoro a potom reflexy z horných končatín.
Pseudohypertrofia sa môže vyvinúť nielen v oblasti gastrocnemia, ale aj v gluteálnych, deltových, brušných a jazykových svaloch. Veľmi často srdcový sval trpí typom kardiomyopatie. Odhalia sa poruchy rytmu srdcovej činnosti, rozšírenie hraníc srdca, hluchota tónov, zmeny na EKG. Akútne srdcové zlyhanie je najčastejšou príčinou smrti pri Duchennovej myodystrofii. Pri pitve sa zistí fibróza a tuková infiltrácia srdcového svalu.
Často dochádza k porušeniu motility gastrointestinálneho traktu.
Bežným príznakom je znížená inteligencia. Zaujímavosťou je, že v niektorých rodinách sa oligofrénia prejavuje ostro, v iných relatívne mierne. Zmena vyšších mentálnych funkcií väčšinou neprogreduje a nekoreluje so závažnosťou svalového defektu. Nedá sa to vysvetliť len pedagogickým zanedbávaním chorých detí, ktoré predčasne odchádzajú z detských kolektívov, nenavštevujú Materská škola a škola z dôvodu motorického postihnutia. CT a MRI často odhalia cerebrálnu atrofiu, pravdepodobne spojenú s narušeným prenatálnym vývojom mozgu.
Často sa u detí vyvinie adiposogenitálny syndróm, niekedy aj iné príznaky endokrinnej insuficiencie. Často sú zistené zmeny na kostrovom systéme: deformácia chodidiel, hrudníka, chrbtice, difúzna osteoporóza.
Výrazná vlastnosť formy DMD je vysoký stupeň hyperenzýmy už v skorých štádiách vývoja procesu. Hladina enzýmu špecifického pre svalové tkanivo - kreatinín fosfokinázy - v krvnom sére teda môže desiatky a dokonca stokrát prekročiť normálne hodnoty. Prudké (10-100-násobné) zvýšenie kreatinínfosfokinázy (CPK) v neuromuskulárnej patológii by malo vyvolať diskusiu predovšetkým o nasledujúcich ochoreniach: Duchennova choroba, Beckerova choroba, poliomyozitída a dermatomyozitída, paroxyzmálna myoglobulinúria, distálna myodystrofia. Až v pokročilých štádiách ochorenia sa stupeň hyperenzýmy postupne znižuje. Existujú správy o zvýšení CPK v štádiu vnútromaternicového vývoja.
Duchennova svalová dystrofia sa prenáša recesívnym spôsobom viazaným na X. Gén sa nachádza na krátkom ramene X chromozómu. Frekvencia génových mutácií je pomerne vysoká (30%), čo vysvetľuje veľké množstvo sporadické prípady.
Mutácia (najčastejšie delécia) vedie k sexuálnej alebo takmer úplnej absencii génového produktu - štruktúrneho proteínu dystrofika. Fyziologická úloha dystrofia nebola úplne preukázaná. Nachádza sa vo vysokých koncentráciách v sarkoléme, zrejme hrá úlohu pri udržiavaní integrity tejto membrány. Absencia dystrofických spôsobuje štrukturálne zmeny v sarkoléme, čo následne vedie k strate intracelulárnych zložiek a zvýšenému príjmu vápnika, čo v konečnom dôsledku vedie k odumretiu myofibríl. Predpokladá sa, že nedostatok dystrofických v synaptických zónach kortikálnych neurónov je príčinou mentálnej retardácie.
Pre lekárske genetické poradenstvo je veľmi dôležité vytvorenie heterozygotného nosiča. Pri Duchennovej svalovej dystrofii u heterozygotov sa v približne 70 % prípadov zistia subklinické a niekedy zjavné príznaky svalovej patológie - určité zhutnenie a dokonca nárast lýtkových svalov, rýchla svalová únava počas cvičenia, zmeny EMG a patomorfologické vyšetrenie svalov bioptické vzorky. Najčastejšie heterozygotní nosiči vykazujú zvýšenie aktivity kreatinínfosfokinázy.
Pri klinickom obraze Duchennovej myodystrofie u žien by sa mala najskôr vylúčiť možnosť anomálie na chromozóme X - Shereshevsky-Turnerov syndróm (XO), Morrisov syndróm (XY) alebo mozaikovitosť u týchto syndrómov.
Duchennova svalová dystrofia, ktorá sa začína rozvíjať už v prenatálnom období, je v podstate vrodená myopatia a možno ju diagnostikovať krátko po narodení vykonaním svalovej biopsie a stanovením aktivity CPK.
Myodystrofia Becker. Spolu s ťažkou, malígnou formou X-viazanej Duchennovej myodystrofie existuje aj benígna forma - Beckerova choroba. Pokiaľ ide o klinické príznaky, je veľmi podobná Duchennovej forme, ale spravidla začína neskôr - vo veku 10-15 rokov, mierne tečie, pacienti zostávajú schopní pracovať dlho, vo veku 20 rokov -30 rokov a neskôr môžu stále chodiť. Plodnosť nie je znížená, takže choroba sa niekedy vyskytuje vo viacerých generáciách rodiny: chorý muž prenáša chorobu na svojho vnuka prostredníctvom svojej dcéry (“efekt starého otca”). Počiatočné príznaky, podobne ako pri Duchennovej chorobe, sa prejavujú slabosťou svalov panvového pletenca, potom proximálnych dolných končatín. Pacienti menia chôdzu, pociťujú ťažkosti pri výstupe do schodov, pri vstávaní z nízkeho sedadla. Charakterizovaná pseudohypertrofiou lýtkových svalov. Retrakcia kalkaneálnych (Achilových) šliach je menej výrazná ako pri Duchennovej chorobe.
S touto formou nie sú žiadne intelektuálne poruchy, kardiomyopatia chýba alebo je mierne vyjadrená.
Rovnako ako u iných myodystrofií viazaných na X, Beckerova forma významne zvyšuje aktivitu CPK, aj keď v menšom rozsahu ako pri DMD, nepresahuje 5000 jednotiek. Gén pre Beckerovu chorobu, podobne ako Duchennova choroba, je lokalizovaný v krátkom ramene X chromozómu; je pravdepodobné, že oba lokusy spolu úzko súvisia alebo sú alelické. Na rozdiel od Duchennovej choroby, pri ktorej prakticky neexistuje dystrofia, sa pri Beckerovej chorobe syntetizuje abnormálna dystrofia. Rozdiely sa nachádzajú aj pri svalovej biopsii. Pri Beckerovej svalovej dystrofii svalové vlákna zvyčajne nie sú okrúhle, hyalínové vlákna, charakteristické pre Duchennovu svalovú dystrofiu, sú extrémne zriedkavé.
Landouzy-Dejerine myodystrofia (myodystrofia tváre a ramien). Ochorenie sa prenáša autozomálne dominantným spôsobom s vysokou penetranciou, ale trochu premenlivou expresivitou. Vyskytuje sa oveľa menej často ako Duchennova myodystrofia (0,4 na 100 tisíc obyvateľov). Predpokladá sa, že gén pre toto ochorenie je lokalizovaný na 4. chromozóme. Ženy ochorejú častejšie ako muži (3:1), fyzické preťaženie, intenzívne športovanie a iracionálne vedené fyzioterapeutické cvičenia môžu prispieť k vážnejšiemu priebehu ochorenia.
Landouzy-Dejerine myodystrofia je relatívne priaznivá súčasná forma svalovej patológie. Začína vo veku okolo 20 rokov, niekedy aj neskôr. Avšak v rodinných prípadoch ochorenia, keď je možné sledovať dynamiku mladších členov rodiny, je možné zistiť určitú slabosť svalov, napríklad svalov tváre, a v skoršom veku .
Svalová slabosť a atrofia sa najskôr objavia v svaloch tváre alebo ramenného pletenca. Postupne sa tieto poruchy rozšírili do svalov proximálnych ramien a potom do dolných končatín. Vo väčšine prípadov sú najskôr postihnuté svaly prednej plochy nôh (s rozvojom visiacej nohy), potom svaly proximálnych nôh. Vo vrchole ochorenia sú výrazne postihnuté kruhové svaly oka a úst, veľký prsný sval, predný pílovitý sval a dolné úseky trapézového svalu, široký chrbtový sval, biceps, triceps ramena. Charakteristický je vzhľad pacientov: typická tvár myopata s „priečnym úsmevom“ („úsmev Giocondy“), výčnelok horná pera(„tapírové pery“), výrazné pterygoidné lopatky, zvláštna deformita hrudníka s jeho sploštením v predozadnom smere a rotáciou vo vnútri ramenných kĺbov. Často dochádza k asymetrii lézie, dokonca aj v rámci jedného svalu (napr. orbicularis oculi). Možno pozorovať pseudohypertrofiu gastrocnemia, deltových svalov a niekedy aj tvárových svalov. Kontraktúry a retrakcie sú vyjadrené stredne. Šľachové reflexy sú dlho zachované, ale niekedy sa znižujú už v ranom štádiu.
Príznaky poškodenia srdcového svalu sú zriedkavé. Aktivita sérových enzýmov je mierne zvýšená a môže byť normálna. Intelekt netrpí. Priemerná dĺžka života vo väčšine prípadov nie je znížená. Zaujímavosťou je, že EMG pri Landouzy-Dejerine myodystrofii často nie je celkom typické pre svalovú úroveň lézie. U niektorých pacientov (členov jednej rodiny) môže dôjsť k zníženiu amplitúdy biopotenciálov, interferenčnému typu krivky, u iných naopak k zníženiu frekvencie a hypersynchrónnej aktivity, niekedy s typickým plotom. rytmus. Malo by sa pamätať na spinálny variant, ktorý napodobňuje Landouzyho-Dejerinovu chorobu.
Erb-Roth myodystrofia (myodystrofia končatín a pletenca). Prenáša sa autozomálne recesívnym spôsobom, obe pohlavia sú rovnako postihnuté. Začiatok ochorenia sa vo väčšine prípadov vzťahuje na polovicu 2. dekády života (14-16 rokov), popisuje sa však ako skorá, pseudo-Duchennova forma, kedy sa prvé príznaky objavia pred 10. rokom života. a ochorenie je ťažké a neskorý variant s nástupom po 30 rokoch.
Priebeh ochorenia môže byť rýchly alebo pomalší, v priemere k úplnej invalidite dochádza do 15-20 rokov od objavenia sa prvých príznakov. Myodystrofia začína buď poškodením svalov panvového pletenca a proximálnych nôh (forma Leiden-Mobius), alebo z pletenca ramenného (forma Erb). V niektorých prípadoch sú ramenný a panvový pletenec postihnuté súčasne. Pomerne výrazne trpia svaly chrbta a brucha. Pacienti majú charakteristickú „kačaciu“ chôdzu, je ťažké vstať z ľahu a sedu, je zdôraznená bedrová lordóza. Svaly tváre nie sú vo väčšine prípadov ovplyvnené. Pre túto formu nie sú charakteristické kontraktúry a pseudohypertrofia. Môžu sa vyskytnúť terminálne atrofie a retrakcie šľachy. Inteligencia je zvyčajne zachovaná. Srdcový sval je väčšinou nedotknutý. Hladina enzýmov v krvnom sére je spravidla zvýšená, ale nie tak prudko ako pri X-viazanej myodystrofii. Existujú náznaky, že u mužov je hladina CPK vyššia ako u žien. Existuje významný rozdiel v expresii mutantného génu u rôznych členov rodiny - spolu so závažným klinickým obrazom môžu byť relatívne mierne a dokonca vymazané klinické príznaky. Smrť zvyčajne nastáva v dôsledku pľúcnych komplikácií.
Keďže klinika myodystrofie končatín je zvlášť ochotná imitovať nervovosvalové ochorenia iného charakteru, je potrebné, najmä v ojedinelých prípadoch a pri neskoršom nástupe ochorenia, vykonať dôkladné klinické vyšetrenie na vylúčenie spinálnej amyotrofie, polymyozitídy, metabolické, endokrinné, toxické, liečivé, karcinomatózne myopatie. V minulosti bola jasná nadmerná diagnóza tejto formy svalovej dystrofie.
Liečba svalových dystrofií. Terapeutické možnosti svalových dystrofií sú veľmi obmedzené. Etiologická a patogenetická liečba prakticky neexistuje. Symptomatická liečba je zameraná predovšetkým na zabránenie vzniku kontraktúr, udržanie existujúcej svalovej sily a prípadne na určité zníženie miery atrofie. Hlavnou úlohou je maximalizovať obdobie, počas ktorého sa pacient môže pohybovať samostatne, pretože v polohe na chrbte sa rýchlo zvyšujú kontraktúry, skolióza a poruchy dýchania. Lekársky komplex by mal zahŕňať terapeutické cvičenia, masáže, ortopedické opatrenia, liekovú terapiu.
Terapeutická gymnastika pozostáva z pasívnych a aktívnych pohybov vykonávaných vo všetkých kĺboch v rôznych polohách: v stoji, v sede, v ľahu, s rôznymi polohami končatín. Aktívne pohyby sa prednostne vykonávajú v izometrickom režime. Gymnastika by sa mala vykonávať pravidelne niekoľkokrát denne. Zároveň treba varovať pred nadmerným cvičením, najmä tým, ktoré je sprevádzané presilením svalov. Dôležité (najmä po imobilizácii pacienta) sú dychové cvičenia.
Zachovať možnosť samostatného pohybu majú za cieľ aj ortopedické opatrenia konzervatívneho (špeciálne pneumatiky) a operačného charakteru (Achilleotómia, transekcia m. gastrocnemius), zamerané na korekciu kontraktúr a vznikajúceho patologického nastavenia končatín. V každom prípade je potrebné individuálne zvážiť očakávaný prínos a možné poškodenie chirurgického zákroku. Malo by sa pamätať na to, že často (najmä pri ťažkej hyperlordóze a slabosti štvorhlavého stehenného svalu) má ekvinovarusová výsadba chodidiel kompenzačný význam a napríklad po achilotómii môže byť pacient úplne imobilizovaný. S rozvíjajúcimi sa kontraktúrami sa odporúča opatrne naťahovať svaly až 20-30 krát denne, po čom nasleduje dlahovanie počas spánku.
Lieková terapia zahŕňa vymenovanie metabolických liekov zameraných na vyplnenie nedostatku energie a bielkovín, ale ich účinnosť je veľmi pochybná. Používajú sa antagonisty vápnika (kvôli defektu bunkových membrán identifikovanom pri Duchennovej chorobe, čo vedie k zvýšenému príjmu vápnika do bunky), imunomodulátory, zlúčeniny obsahujúce fosfor (ATP, fosfaden), vitamín E (100 mg perorálne 3-krát ročne). deň). Ukázalo sa, že užívanie prednizolónu (0,75 mg/kg denne) pri DMD môže dramaticky zvýšiť svalovú silu, ale tento účinok pretrváva nie dlhšie ako rok a vo všeobecnosti neovplyvňuje výsledok ochorenia. Vzhľadom na závažné vedľajšie účinky, ktoré sa vyskytujú pri dlhodobom užívaní lieku, sa jeho užívanie neodporúča. Odhady účinku anabolických steroidov sú rozporuplné a ich vymenovanie je často spojené s neopodstatneným rizikom. Pri hodnotení účinku niektorých liekov pri DMD treba mať na pamäti, že pri strednej závažnosti ochorenia u pacientov vo veku 3-6 rokov môže dôjsť k relatívnej stabilizácii stavu súvisiaceho s vekom podmieneným vývojom svalovej systém, osvojenie si pohybových schopností, ktoré môžu do určitej miery dočasne kompenzovať prebiehajúci dystrofický proces.
Určitá dôležitá je úprava výživy pacienta, odporúča sa diéta s vysokým obsahom bielkovín a nízkym obsahom tukov a zníženým obsahom kalórií s optimálnym obsahom vitamínov a mikroelementov. Dôležitú úlohu zohráva psychická podpora pacienta, pokračovanie vo vzdelávaní, správna profesijná orientácia.
Pri niektorých ochoreniach sú ovplyvnené kontrakcie kostrového svalstva. V mnohých prípadoch sú porušenia spôsobené patologickým stavom nie samotných svalových vlákien, ale zodpovedajúcich častí nervového systému. Napríklad poliomyelitída, vírusová infekcia, ktorá ničí motorické neuróny, spôsobuje paralýzu kostrových svalov a dokonca smrť v dôsledku zlyhania dýchania. Celková hmotnosť kostrových svalov je až 40% telesnej hmotnosti. V ľudskom tele je až 400 svalov, ktoré pozostávajú z kostrových svalové tkanivo. Kostrové svaly- orgány, ktoré vykonávajú hlavnú funkciu pohybu. Medzi doplnkovými funkciami svalov stojí za zmienku účasť svalov na návrate periférnej krvi do srdca, najmä táto doplnková funkcia je vyjadrená vo svaloch dolných končatín. Okrem toho v podmienkach hypotermie svaly vykonávajú kalorickú funkciu.
Medzi ochoreniami kostrového svalstva sú najčastejšie ochorenia priečne pruhovaného svalstva dystrofického (myopatia) a zápalového charakteru (myozitída). Svaly môžu byť zdrojom množstva nádorov. Medzi myopatiami sú obzvlášť zaujímavé progresívna svalová dystrofia (progresívna myopatia) a myopatia pri myasthenia gravis.
Progresívna svalová dystrofia (progresívna myopatia) zahŕňa rôzne primárne dedičné chronické ochorenia priečne pruhovaného svalstva (nazývajú sa primárne, pretože nedochádza k poškodeniu miechy a periférnych nervov). Ochorenie je charakterizované rastúcou, zvyčajne symetrickou, svalovou atrofiou, sprevádzanou progresívnou svalovou slabosťou až po úplnú nehybnosť.
Etiológia a patogenéza málo študovaný. Diskutuje sa o význame anomálií v štruktúrnych proteínoch, sarkoplazmatickom retikule, inervácii, enzymatickej aktivite svalových buniek. Charakteristické je zvýšenie aktivity svalových enzýmov v krvnom sére, zodpovedajúce elektrofyziologické poruchy v poškodených svaloch a kreatinuria.
Klasifikácia. V závislosti od typu dedičnosti, veku, pohlavia pacientov, lokalizácie procesu a priebehu ochorenia sa rozlišujú 3 hlavné formy progresívnej svalovej dystrofie: Duchenne, Erb a Leucene. Morfologické charakteristiky týchto foriem svalovej dystrofie sú podobné.
Duchennova svalová dystrofia (skorá forma) s recesívnym typom dedičnosti spojenej s chromozómom X sa zvyčajne objavuje vo veku 3-5 rokov, častejšie u chlapcov. Najprv sú postihnuté svaly panvového pletenca, stehien a nôh, potom ramenného pletenca a trupu. Erbova svalová dystrofia (juvenilná forma) má autozomálne dominantný typ dedičnosti, vyvíja sa počas puberty. Postihnuté sú najmä svaly hrudníka a ramenného pletenca, niekedy aj tvár (myopatická tvár – hladké čelo, nedostatočné zatvorenie očí, hrubé pery). Možná atrofia svalov chrbta, panvového pletenca, proximálnych končatín. Autozomálne recesívna Leidenská svalová dystrofia začína v detstve alebo puberte a postupuje rýchlejšie ako juvenilná forma (Erba), ale priaznivejšie ako skorá forma (Duchene). Proces, počnúc svalmi panvového pletenca a bokov, postupne zachytáva svaly trupu a končatín.
Patologická anatómia. Svaly sú zvyčajne atrofické, stenčené, ochudobnené o myoglobín, preto na reze pripomínajú rybie mäso. Svalový objem sa však môže zväčšiť aj vďaka voľnému rastu tukového tkaniva a spojivového tkaniva, čo je charakteristické najmä pre Duchennovú svalovú dystrofiu (pseudohypertrofická svalová dystrofia).
Pri mikroskopickom vyšetrení majú svalové vlákna rôzne veľkosti: spolu s atrofickými sú ostro zväčšené, jadrá sú zvyčajne umiestnené v strede vlákien. Vyjadrujú sa dystrofické zmeny svalových vlákien (akumulácia lipidov, zníženie obsahu glykogénu, vymiznutie priečneho pruhovania), ich nekróza a fagocytóza. V jednotlivých svalových vláknach sa zisťujú známky regenerácie. Tukové bunky sa hromadia medzi poškodenými svalovými vláknami. Pri ťažkom priebehu ochorenia sa medzi rozsiahlymi výrastkami tukového a spojivového tkaniva nachádzajú iba jednotlivé atrofické svalové vlákna.
Ultraštrukturálne zmeny vo svalových vláknach boli podrobnejšie študované pri Duchennovej svalovej dystrofii. Na začiatku ochorenia sa zistí expanzia sarkoplazmatického retikula, ložiská deštrukcie myofibríl, rozšírenie medzifibrilárnych priestorov, v ktorých sa zvyšuje množstvo glykogénu a pohyb jadier do stredu vlákna. V neskorom štádiu ochorenia myofibrily podliehajú fragmentácii a dezorganizácii, mitochondrie sú opuchnuté, T-systém je rozšírený; vo svalových vláknach sa zvyšuje počet lipidových inklúzií a glykogénu, objavujú sa autofagolyzozómy. Na konci choroby sa svalové vlákna stávajú hustejšími, obklopené látkou podobnou hyalínom, okolo nekrotických svalových vlákien sa objavujú makrofágy a tukové bunky.
Smrť u pacientov s ťažkou progresívnou svalovou dystrofiou sa spravidla vyskytuje pľúcna infekcia.
Choroby spôsobené porušením energetický metabolizmus vo svaloch
V kostrovom svalstve sa zvyčajne využívajú dva hlavné zdroje energie – mastné kyseliny a glukóza. Preto môže byť narušenie využitia glukózy alebo tukov sprevádzané jasnými klinickými prejavmi na strane svalového systému. Najzávažnejším prejavom tejto patológie je syndróm akútnej bolesti svalov, ktorý môže viesť k závažnej rabdomyolýze a myoglobinúrii. Treba spomenúť aj progresívnu svalovú slabosť simulujúcu svalovú dystrofiu. Neexistuje žiadne vysvetlenie pre existenciu týchto dvoch odlišných klinických syndrómov.
Glykogenóza (ochorenie ukladania glykogénu) a glykolytické defekty. Rozlišujú sa štyri typy porúch metabolizmu glykogénu (typy II, III, IV a V) a štyri typy porúch glykolýzy (typy VII, IX, X a XI), ktoré sa prejavujú výraznými poruchami kostrového svalstva
Nedostatok kyslej maltázy (glykogenóza typu II). Kyslá maltáza je lyzozomálny enzým zo skupiny kyslých hydroláz, ktorý má a-1,4 a a-1,6 glukozidázovú aktivitu: rozkladá glykogén na glukózu. Zároveň nie je jasne definovaná úloha tohto enzýmu v metabolizme uhľohydrátov. Existujú tri klinické formy deficitu kyslej maltázy, z ktorých každá je dedená autozomálne recesívnym spôsobom. Biochemický základ rôznych klinických prejavov tohto nedostatku enzýmu je nejasný.
V dojčenskom veku sa nedostatok kyslej maltázy prejavuje ako celková glykogenóza. Pri narodení nie je zistená žiadna patológia, ale čoskoro sa zistí ostrá svalová slabosť, kardiomegália, hepatomegália a výrazné zvýšenie veľkosti jazyka. Akumulácia glykogénu v motorických neurónoch miechy, ako aj v mozgovom kmeni, zhoršuje svalovú slabosť. Takéto deti zvyčajne zomierajú do prvého roku života.
U detí a dospelých sa toto ochorenie prejavuje ako svalová dystrofia. Detské formy ochorenia sú charakterizované pomalým vývojom dieťaťa, slabosťou proximálnych svalov končatín a zväčšením veľkosti lýtkových svalov. Choroba môže progredovať s rozvojom respiračného zlyhania; smrť nastáva spravidla na konci 2. dekády života. Môže sa vyskytnúť srdcové postihnutie, ale hepatomegália a makroglosia sú zriedkavé.
Ochorenie u dospelých začína v 3. – 4. dekáde života a môže byť chybne diagnostikované ako dystrofia končatín a pletenca alebo polymyozitída. Počiatočným prejavom ochorenia je zlyhanie dýchania v dôsledku slabosti bránice. Pečeň, srdce a jazyk zvyčajne nie sú ovplyvnené. Predpoklad diagnózy vzniká po štúdiu svalovej biopsie, v ktorej sa nachádzajú vakuoly obsahujúce glykogén a kyslú fosfatázu. Elektrónová mikroskopia ukazuje, že glykogén je spojený s membránami a je voľne umiestnený v tkanivách. Konečná diagnóza je stanovená biochemickým vyšetrením postihnutého svalu. Aktivita kyslej maltázy v moči je znížená. Úroveň aktivity CK v sére môže prekročiť normu 10-krát. Pomocou EMG je možné odlíšiť deficit maltázy od svalovej dystrofie vysokofrekvenčnými myotonickými výbojmi sprevádzajúcimi krátkodobé potenciály motorických jednotiek na pozadí fibrilácií a pozitívnych vrcholových potenciálov.
Nedostatok enzýmu, ktorý inhibuje vetvenie molekuly glykogénu (glykogenóza typu III). Toto pomerne ľahké detské ochorenie sa prejavuje hepatomegáliou, retardáciou rastu a hypoglykémiou; neostro vyjadrená svalová slabosť sa pozoruje zriedkavo. Po puberte sa tieto príznaky zvyčajne zmiernia alebo úplne vymiznú, takže svalová slabosť a určitá strata svalovej hmoty môžu byť jednoducho spôsobené znížením cvičenia v dôsledku zlej tolerancie cvičenia. Predpoklad možnej diagnózy vzniká vtedy, keď po tom, čo pacient vykoná špeciálny cvik na svaly predlaktia, sa obsah kyseliny mliečnej v krvi nezvýši. Aktivita CK v sére je zvyčajne zvýšená. EMG odhalí zmeny charakteristické pre myopatiu, ako aj známky zvýšenej dráždivosti membrán myotonickými impulzmi. Svalová biopsia odhalí vakuoly so zvýšeným obsahom glykogénu. Na potvrdenie diagnózy je potrebná biochemická štúdia svalu.
Nedostatok enzýmu vetviaceho glykogén (glykogenóza typu IV). Nedostatok tohto enzýmu je veľmi ťažká, smrteľná patológia dojčenského veku, pri ktorej poruchy kostrového svalstva ustupujú do pozadia v porovnaní s rozvojom chronického zlyhania pečene. Svalová hypotónia a svalová atrofia však môžu naznačovať primárne svalové ochorenie alebo spinálnu svalovú atrofiu.
Nedostatok svalovej fosforylázy (glykogenóza typu V). Zlá tolerancia cvičenia je charakteristickým príznakom nedostatku svalovej fosforylázy, prvýkrát opísaný v roku 1951. McArdle. Ochorenie sa dedí autozomálne recesívnym spôsobom; muži ochorejú častejšie ako ženy. Po puberte pacienti pociťujú bolestivé svalové kŕče a rýchlu svalovú únavu po intenzívnej fyzickej aktivite – behu, zdvíhaní závažia. V literatúre sú opísané varianty ochorenia, začínajúce v detstve aj neskôr. Mnohí pacienti uvádzajú fenomén druhého dychu, ktorý nastáva po krátkom odpočinku alebo po spomalení tempa fyzickej aktivity, čo im umožňuje udržať si pohybovú aktivitu po mnoho rokov. Fyzická prepracovanosť u takýchto pacientov vedie k rozvoju rabdomyolýzy, myoglobinúrie a zlyhania obličiek. Trvalá svalová slabosť a progresívna svalová atrofia sú zriedkavé, takže ich fyzické vyšetrenie v obdobiach medzi exacerbáciami ochorenia zvyčajne neodhalí patológiu. Ostatné orgány pri tejto chorobe nie sú ovplyvnené.
Aktivita CK v sére podlieha významným výkyvom a môže byť zvýšená aj počas asymptomatických období. Test so zaťažením svalov predlaktia nie je sprevádzaný zvýšením obsahu kyseliny mliečnej v krvi. EMG nálezy sú normálne, pokiaľ sa nevykonajú bezprostredne po epizóde rabdomyolýzy. Svalová biopsia odhalí vezikuly obsahujúce glykogén pod sarkolemou. Prítomnosť nedostatku svalovej fosforylázy môže byť preukázaná histochemickým farbením histologickej vzorky alebo biochemickým vyšetrením svalového tkaniva. Pacienti môžu zostať dosť aktívni počas celého života za predpokladu, že sa zdržia určitých fyzických preťažení. Substitučná diétna terapia glukózou alebo fruktózou zvyčajne nie je sprevádzaná oslabením príznakov ochorenia.
Nedostatok fosfofruktokinázy (glykogenóza typu VII). Táto porucha sa podobá nedostatku svalovej fosforylázy a je tiež dedená autozomálne recesívnym spôsobom; medzi chorými prevládajú muži. Rovnako ako pri nedostatku fosforylázy, provokatívnych momentoch a laboratórnych údajoch. Tento typ nedostatku enzýmu sa zisťuje histochemickým farbením svalového preparátu na fosfofruktokinázu (FFK). Pre spoľahlivú diagnózu je potrebná biochemická štúdia svalových enzýmov. U niektorých pacientov s nedostatkom tohto enzýmu je možná mierna hemolýza, zvýšenie počtu retikulocytov v periférnej krvi a zvýšenie obsahu bilirubínu v krvi, pretože nedostatok FGFK sa vyskytuje nielen vo svaloch, ale aj v erytrocytoch.
Syndrómy spojené s nedostatočnosťou nového glykolytického enzýmu. Od roku 1981 boli identifikované tri ďalšie nedostatky glykolytických enzýmov: fosfoglycerátkináza (PGlkK) (typ IX), fosfoglycerátmutáza (PGLM) (typ X) a laktátdehydrogenáza (LDH) (typ XI). Klinický obraz všetkých týchto troch typov enzýmového deficitu je identický. V ranom detstve alebo adolescencii, po fyzickom nadmernom zaťažení, pacienti pociťujú epizódy myoglobinúrie a myalgie. Zdá sa, že všetky tieto defekty enzýmov sa dedia autozomálne recesívnym spôsobom. Aktivita CK v sére sa môže zvýšiť tak počas exacerbácií ochorenia, ako aj medzi exacerbáciami. Pri nedostatočnosti FGLM a LDH je zvýšenie obsahu kyseliny mliečnej v krvi po záťaži svalov predlaktia zvyčajne nižšie ako normálne. Pri nedostatku FGlK sa hladina laktátu v krvi po cvičení vôbec nezvyšuje. Vo všeobecnosti je táto forma enzýmového deficitu v klinických prejavoch veľmi podobná nedostatočnosti svalovej fosforylázy a fosfofruktokinázy. Histologické vyšetrenie svalov pri týchto formách enzýmového deficitu býva neinformatívne, dochádza len k miernemu zvýšeniu obsahu glykogénu vo svaloch. Pre spoľahlivú diagnózu je potrebná biochemická štúdia svalu.
Voľné mastné kyseliny ako zdroj energie sa tvoria z triglyceridov nahromadených vo svale a z cirkulujúcich lipoproteínov s veľmi nízkou hustotou, ktoré sa vplyvom endoteliálnej lipoproteínovej lipázy v kapilárach rozkladajú. Karnitín, nevyhnutný substrát pre metabolizmus lipidov, sa tvorí v pečeni a transportuje sa do svalu. Vo svaloch sa voľné mastné kyseliny kombinujú s koenzýmom A (CoA-SH) pod vplyvom mastnej acylsyntetázy nachádzajúcej sa vo vonkajšej mitochondriálnej membráne, čo vedie k tukovému acylkoenzýmu A (F-acyl-CoA). Transport cez mitochondriálnu vnútornú membránu vyžaduje prenos karnitínu cez karnitín palmitínový transfúzny enzým I (CPT-1) naviazaný na vonkajší povrch mitochondriálnej vnútornej membrány. V mitochondriách sa tukový acylkarnitín (P-acylkarnitín) syntetizuje pomocou CPT-P, ktorý sa viaže na vnútorný povrch vnútornej mitochondriálnej membrány. V tomto prípade mastný acylkoenzým A podlieha b-oxidácii.
poruchy metabolizmu lipidov. Lipidy sú dôležitým energetickým substrátom najmä počas svalového odpočinku a pri dlhšej, nie však prudkej fyzickej námahe.
Nedostatok karnitínu. Existujú myopatické a systémové (generalizované) formy nedostatku karnitínu.
Myopatický nedostatok karnitínu sa zvyčajne prejavuje všeobecnou svalovou slabosťou, ktorá zvyčajne začína v detstve. Klinické prejavy tohto ochorenia čiastočne pripomínajú svalovú dystrofiu a čiastočne polymyozitídu. Väčšina prípadov je sporadická; verí, že choroba môže byť dedená autozomálne recesívnym spôsobom. Niekedy sa vyskytuje kardiomyopatia. Aktivita CK v sére je mierne zvýšená; na EMG - známky myopatie. Svalová biopsia odhalí výraznú akumuláciu lipidov. Obsah karnitínu v krvnom sére je normálny. Predpokladá sa, že toto ochorenie narúša transport karnitínu do svalov, preto je jeho obsah vo svaloch taký nízky. Niektorí pacienti reagujú na perorálnu substitučnú terapiu karnitínom pozitívne, v každom prípade ju treba vyskúšať vo všetkých prípadoch. Iní pacienti z plesových sál reagovali pozitívne na liečbu prednizolónom z neznámych dôvodov. U niektorých pacientov malo nahradenie triglyceridov so stredne dlhým reťazcom triglyceridmi s dlhým reťazcom v ich strave terapeutický účinok. Niektorí pacienti dobre reagujú na liečbu riboflavínom.
Systémový nedostatok karnitínu je autozomálne recesívne ochorenie detstva a raného detstva. Je charakterizovaná progresívnou svalovou slabosťou a epizódami hepatálnej encefalopatie s nevoľnosťou, vracaním, stratami vedomia, kómou a predčasnou smrťou. Nízke hladiny karnitínu v sére odlišujú túto formu od nedostatku myopatického karnitínu. Nebola identifikovaná žiadna príčina, ktorá by mohla spôsobiť alebo vysvetliť nízky obsah karnitínu v krvi. U niektorých pacientov je zistená znížená syntéza karnitínu, u iných - jeho zvýšené vylučovanie močom. Aktivita CK v sére môže byť mierne zvýšená. Vo svalovej biopsii sa zistí akumulácia lipidov. V niektorých prípadoch je ich akumulácia zaznamenaná aj v pečeni, srdci a obličkách. U niektorých pacientov, ale nie u všetkých, bol účinný perorálny karnitín alebo kortikosteroidy.
Nedostatok karnitín palmityltransferázy. Tento nedostatok enzýmu sa prejavuje opakovanou myoglobinúriou. Nie je presne známe, ktorá aktivita karnitín palmitín transferázy (CPT) v tomto prípade klesá: CPT-I alebo CPT-II. Zdá sa, že tento nedostatok enzýmu je výsledkom dysregulácie abnormálnych vlastností enzýmu. Veľká fyzická aktivita (hranie futbalu, dlhá túra) môže vyvolať rabdomyolýzu; niekedy však nie je možné identifikovať provokujúci faktor. Prvé príznaky ochorenia sa často objavujú v detstve. Na rozdiel od svalového poškodenia pri poruchách glykolýzy, kedy sa po krátkodobej, ale intenzívnej fyzickej námahe objavia svalové kŕče, kvôli ktorým pacient odmieta pokračovať vo fyzickej aktivite a tým sa chráni, pri nedostatku CBT sa svalová bolesť nedostaví, kým sa nevyčerpajú všetky energetické zdroje sval je vyčerpaný a jeho deštrukcia nezačne. Počas rabdomyolýzy dochádza k silnej svalovej slabosti, takže niektorí pacienti môžu potrebovať mechanickú ventiláciu. Na rozdiel od deficitu karnitínu, pri deficite CBT medzi záchvatmi je svalová sila zachovaná a svalová biopsia neodhalí akumuláciu lipidov v nej. Diagnóza vyžaduje priame vyšetrenie obsahu CPT vo svale. Liečba pozostáva zo zvýšenia príjmu sacharidov v potrave pred cvičením alebo nahradenia triglyceridov so stredne dlhým reťazcom triglyceridmi s dlhým reťazcom v strave pacienta. Všetky tieto liečby však nie sú úplne uspokojivé.
Nedostatok myoadenylátdeaminázy. Enzým adenylátdeamináza premieňa 5-adenozínmonofosfát (5-AMP) na inozínmonofosfát (IMP) s uvoľňovaním amoniaku, ktorý môže hrať úlohu pri regulácii svalového adenozíntrifosfátu (ATP). V roku 1978 podarilo identifikovať skupinu pacientov so svalovou bolesťou a intoleranciou záťaže, ktorí mali deficit izoenzýmu myoadenylátdeaminázy. Nedostatok tohto enzýmu je pomerne častý a vyskytuje sa približne u 1 % populácie, čo sa dá zistiť špeciálnym farbením svalových histologických preparátov alebo biochemickým vyšetrením svalového tkaniva. Pri skúmaní testu so zaťažením svalov predlaktia sa zistí zníženie tvorby amoniaku. Od pôvodného popisu tohto ochorenia nebolo možné identifikovať jeho jasnejšie klinické prejavy. Nedostatok tohto enzýmu často vykazujú aj pacienti s inými neuromuskulárnymi patológiami (poškodenie buniek predných rohov miechy, svalová dystrofia, myasthenia gravis). Presný %��D0�indikatívny význam tohto porušenia nebol stanovený.
Mitochondriálne myopatie. Heterogénna skupina ochorení charakterizovaná mitochondriálnou patológiou vďačí za svoj názov špeciálnemu typu trichrómom zafarbeného histologického preparátu bioptického svalu. Kearns-Sayreov syndróm je sporadické ochorenie, ktoré začína v detstve a je charakterizované progresívnou vonkajšou oftalmoplégiou, poruchami intrakardiálneho vedenia, ktoré často vedie k úplnej priečnej blokáde. Zaznamenáva sa aj degenerácia sietnice, malý vzrast pacientov, gonádové defekty.
Dedičná porucha s progresívnou vonkajšou oftalmoplégiou a slabosťou proximálneho svalstva môže byť ťažké odlíšiť od Kearns-Sayrovho syndrómu. Nedávno bol identifikovaný ďalší syndróm, označený skratkou MERRF 1 , pri ktorom sa myoklonická forma epilepsie kombinuje s hrubými červenými vláknami nachádzajúcimi sa v histologických preparátoch svalov. Ochorenie sa vyskytuje medzi 1. a 5. dekádou života a je charakterizované generalizovanými záchvatmi, myoklonom, demenciou, stratou sluchu a ataxiou.
Tretím ochorením z tejto skupiny je syndróm MELAS 2 (1 MERRF - myotonicepilepsia, ragged-redfibers (pozn. red.). 2 MELAS-myopathyencefalopatia, laktikacidóza, epizódy podobné mŕtvici (pozn. red.), čo je pomaly progresívne ochorenie charakterizované mitochondriálnou myopatia, encefalopatia, laktátová acidóza, epizódy podobné mŕtvici s prechodnou hemiparézou, hemianopsiou alebo kortikálnou slepotou a fokálnymi alebo generalizovanými záchvatmi namiesto chromozomálnej DNA.
Strana 44 zo 44
Kostrové svaly sa podieľajú na patologickom procese pri rôznych degeneratívnych, metabolických a zápalových ochoreniach. Vo väčšine prípadov to vedie k degenerácii svalových vlákien a pri chronických formách k ich výmene. spojivové tkanivo a tuku. Proximálne svalové skupiny sú poškodené výraznejšie ako distálne, ako aj dolné končatiny vo vzťahu k horným. Choré dieťa sa vyznačuje takzvanou kačacou (koláčavou) chôdzou, nie je schopné behať, stúpať po schodoch a vstávať, ak je v sede. Jeho šľachové reflexy sú utlmené, stupeň ich vyhasnutia je úmerný stupňu oslabenia svalovej sily. Citlivosť nie je ovplyvnená.
Medzi diagnosticky cenné laboratórne metódy patrí stanovenie aktivity enzýmov, najmä kreatínfosfokinázy, v sére. Tento enzým, ktorý katalyzuje reakciu: fosfokreatín + ADP-kreatín + ATP, je prítomný hlavne v mozgových bunkách a svalovom tkanive. Pri niektorých difúznych svalových ochoreniach, najmä svalovej dystrofii, preniká jeho nadbytočné množstvo do medzibunkového priestoru a krvi. U pacientov je aktivita sérovej laktátdehydrogenázy a glutamín-oxalooctovej transaminázy zvyčajne zvýšená, ale ich široká distribúcia v iných tkanivách, vrátane pečene, znižuje špecificitu testu. Zvyčajne je na objasnenie diagnózy potrebná biopsia svalového tkaniva.
Zápalové ochorenia svalov. Zápal svalového tkaniva sprevádza niektoré infekcie, najmä trichinelózu, toxoplazmózu a tie, ktoré spôsobuje vírus Coxsackie. Často je súčasťou kolagénových ochorení, vrátane dermatomyozitídy, lupus erythematosus, periarteritis nodosa a reumatoidnej artritídy.
Polymyozitída. Difúzny izolovaný zápal svalov neznámej etiológie sa nazýva polymyozitída. Je charakterizovaná rýchlo progresívnym priebehom, slabosťou a bolesťou v proximálnych svalových skupinách. Často sú do procesu zapojené svaly krku, a preto je pre dieťa ťažké zdvihnúť hlavu a udržať ju v tejto polohe. Laboratórne príznaky zápalu svalov zahŕňajú zvýšenie ESR a počtu leukocytov. Ich absencia však nevylučuje polymyozitídu. Hladiny enzýmov v sére sú zvyčajne zvýšené. Pri svalovej biopsii sa zisťuje degenerácia a čiastočná regenerácia vlákien a ich infiltrácia lymfoidnými bunkami. Je ťažké odlíšiť polymyozitídu od svalovej dystrofie a dermatomyozitídy. Môže predstavovať atypickú formu dermatomyozitídy, hoci histológia týchto dvoch stavov je trochu odlišná: dermatomyozitída je charakterizovaná vaskulitídou, ktorá pri polymyozitíde zvyčajne chýba. Prognóza druhého z nich je o niečo priaznivejšia. Liečba kortikosteroidmi je sprevádzaná účinkom, ale pri ich zrušení môže dôjsť k relapsu.
Progresívna osifikujúca myozitída. Etiológia tohto zriedkavého ochorenia spojivového tkaniva a svalov nie je známa. Uvádza sa, že ňou trpia súrodenci vrátane dvojčiat a v priamej línii sa prenáša na pokrvných príbuzných. Predpokladá sa, že choroba sa dedí autozomálne dominantným spôsobom. Chlapci ochorejú 2-3 krát častejšie ako dievčatá.
Patologické príznaky závisia od štádia ochorenia. V počiatočných štádiách sa vo svaloch a šľachách nachádza lokálny edém a infiltráty zápalových buniek. Neskôr sú oblasti zápalu nahradené granulačným tkanivom a nakoniec sa v léziách vytvoria oblasti chrupavkového a kostného tkaniva.
Takmer 75 % chorých detí má vrodené vývojové chyby, najčastejšie nedostatočný vývoj prstov na rukách a ankylózu článkov prstov na nohách a nedostatočný vývoj prvých prstov, polydaktýliu, zakrivenie prstov na rukách, syndaktýliu (nohy), deformáciu ušníc, hluchotu. , absencia zubov. Rovnaké vrodené chyby môžu byť u príbuzných pacienta, u ktorých sa nerozvinulo progresívne ochorenie spojivového tkaniva a svalov. Vek, v ktorom sa myositis ossificans môže začať, sa líši od narodenia po staršie detstvo. Zvyčajne sa rozlišujú tri jeho štádiá: 1) na miestach drobných lokálnych poranení ohraničené, často teplé a mäkké na dotyk, vznikajú pastovité opuchy mäkkých tkanív; 2) po niekoľkých dňoch príznaky zápalu zmiznú a lézia stvrdne; 3) dochádza k osifikácii postihnutej oblasti. Pravidelne sa objavujú nové lézie, najmä na krku a chrbte. Primárnym príznakom môže byť torticollis, ak sa proces vyvinul v sternocleidomastoideus sval. Nakoniec sa osifikácia rozširuje na mnohé šľachy a väzy. Nastupuje ankylóza chrbtice a kĺbov rúk a nôh (obr. 21-5). Zápal sa môže rozšíriť do temporomandibulárnych kĺbov, čo sťažuje žuvacie pohyby. Kostné výrastky môžu vyčnievať cez kožu. V dospievaní choroba často vedie k úplnej imobilizácii a smrti v dôsledku zlyhania dýchania a zastavenia dýchania, hoci existujú správy o prípadoch prežitia. Pri osifikujúcej myozitíde existuje vysoké riziko vzniku osteogénneho sarkómu.
Ryža. 21-5. Dieťa s progresívnou myositis ossificans (typické držanie tela so stuhnutím krku a chrbta).
Niekedy je patologický proces obmedzený na miesto predchádzajúceho poranenia mäkkých tkanív (miositis ossificans circumscripta). Pri chronickej polymyozitíde a dermatomyozitíde sa môže vyskytnúť aj rozšírená kalcifikácia svalového tkaniva.
Výsledky laboratórnych výskumných metód nemajú žiadnu diagnostickú hodnotu.
Hladiny vápnika, fosforu, alkalickej fosfatázy v sére, ako aj aktivita kreatínfosfokinázy a iných enzýmov zostávajú normálne. Kostné tkanivo v lézii sa štruktúrou nelíši od normy.
Existujúce metódy liečby sú neuspokojivé. V niektorých prípadoch sa pri užívaní ACTH a iných kortikosteroidov zaznamenalo spomalenie vývoja ochorenia. Ich úloha v konečnom výsledku liečby je otázna.
Endokrinné a metabolické myopatie. Myopatia pri hypertyreóze je pomerne zriedkavá komplikácia. Je charakterizovaná ptózou, bilaterálnou parézou tvárových svalov a svalov proximálnych končatín. Niektoré príznaky hypertyreózy však môžu byť maskované svalovou slabosťou, no tachykardia, zvýšené potenie a zväčšenie štítnej žľazy zostávajú. Šľachové reflexy, na rozdiel od mnohých iných foriem myopatie, zostávajú normálne. Po úprave hypertyreózy svalová slabosť postupne mizne.
Myopatia pri hypotyreóze. Hypotyreóza u dojčiat môže byť spojená so svalovou slabosťou a hypotenziou. U starších detí s myxedémom sa svalové kontrakcie a relaxácia spomaľujú, v niektorých prípadoch je zaznamenaná svalová hypertrofia (Debre-Semelenov syndróm). Kombinácia znakov, ako je svalová slabosť a hypertrofia, naznačuje svalovú dystrofiu.
Myopatia počas liečby kortikosteroidmi. Môže skomplikovať Itsenko-Cushingovu chorobu, ale častejšie sa vyvíja pri liečbe veľkých dávok syntetických steroidov. Slabosť je badateľná najmä vo svaloch panvového pletenca, čo sa prejavuje kolísavou (kačacou) chôdzou, ťažkosťami pri výstupe do schodov a snahou vstať zo sedu. Trhnutie kolenom chýba. Môže dôjsť k rednutiu svalov. Myopatické zmeny v svalovom tkanive sú zvyčajne bezvýznamné aj pri silnej slabosti. Svalová sila po vysadení kortikosteroidov sa obnovuje pomaly (v priebehu niekoľkých mesiacov).
Myopatia pri hyperparatyreóze. Hyperparatyreóza môže byť spojená so slabosťou a hyporeflexiou v dôsledku hyperkaliémie. Po paratyreoidektómii zvyčajne rýchlo vymiznú.
Nedostatok karnitínu (lipidová myopatia) sprevádza hromadenie veľkého množstva lipidov vo svaloch a v dôsledku toho narušenie ich zásobovania energiou. Karnitín je jednou zo základných zložiek systému, ktorý zabezpečuje prenos mastných kyselín s dlhým reťazcom z cytosolu do mitochondrií, kde dochádza k ich oxidácii. Svalová slabosť sa vyvíja v dvoch formách nedostatku karnitínu.
Nedostatok karnitínu vo svaloch je klinicky reprezentovaný progresívnou slabosťou ich proximálnych skupín, častejšie u školákov a dospievajúcich. Niekedy slabosť prerušovaná a kombinovaná s myoglobinúriou. V závažných prípadoch môže dôjsť k paralýze dýchacích svalov. Sérové hladiny enzýmov (kreatínkinázy a aldolázy) sú zvýšené. Elektromyogram odhaľuje nešpecifické zmeny charakteristické pre myopatiu. Vo svalovej biopsii môžete vidieť veľké množstvo kvapiek tuku. Hladina karnitínu v sére sa nemení, ale vo svaloch klesá. Rozpoznanie patológie je nevyhnutné, pretože sa dá vyliečiť. Často sa mylne považuje za svalovú dystrofiu. Účinok sa môže dostaviť po perorálnom podaní 100 mg / (kg / deň) karnitínu. V niektorých prípadoch je účinná liečba kortikosteroidmi.
- Systémový nedostatok karnitínu sa prejavuje progresívnou myopatiou, vrátane kardiomyopatie, a dysfunkciou pečene, sprevádzané klinikou hepatálnej encefalopatie, ako je Reyeov syndróm. Karnitínová insuficiencia sa od posledne menovaného líši v rekurentnom priebehu a výraznej svalovej slabosti, ktorá pretrváva medzi obdobiami exacerbácie encefalopatie. Hladina kreatínfosfokinázy v sére je výrazne zvýšená, množstvo karnitínu je znížené ako v sére, tak aj vo svaloch. Zmeny v biopsii sú podobné ako pri nedostatku karnitínu vo svalovom tkanive. Podobné klinické a morfologické zmeny, vrátane deficitu karnitínu, možno zistiť pri porušení metabolizmu organických kyselín, napríklad pri metylmalónovej a glutárovej acidúrii (sekundárny deficit karnitínu).
Ryža. 21-6. Dieťa s vrodenou absenciou ľavého prsného svalu. 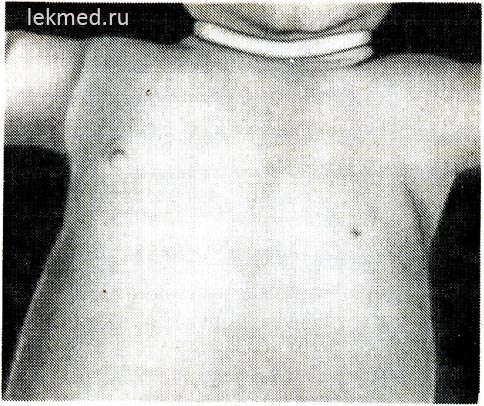
Všimnite si absenciu predného axilárneho záhybu a nízko položenej bradavky.
Liečba spočíva v udržiavaní pacienta na diéte bohatej na sacharidy a nízkym obsahom tukov a v užívaní karnitínu v dennej dávke 100 mg/kg.
Vrodené chyby svalov. Vrodená absencia svalov. Nedostatočný rozvoj svalov môže byť celkom bežný a viesť k úplnej blokáde pohybov kĺbov alebo vrodenej artrogrypóze. Ako vrodená chyba najčastejšie chýba jeden sval. Pomerne častou anomáliou je absencia sternálnej časti m. pectoralis major (obr. 21-6), v niektorých prípadoch sa tento defekt kombinuje so syndaktýliou na postihnutej strane (Poľský syndróm). Absencia prsného svalu často sprevádza svalovú dystrofiu. Vrodená absencia brušných svalov brucha je často spojená s chybami vo vývoji močových ciest.
Ryža. 21-7. Deformácia krku a asymetria tváre u chlapca s vrodenou torticollis, neliečená od 12 rokov. 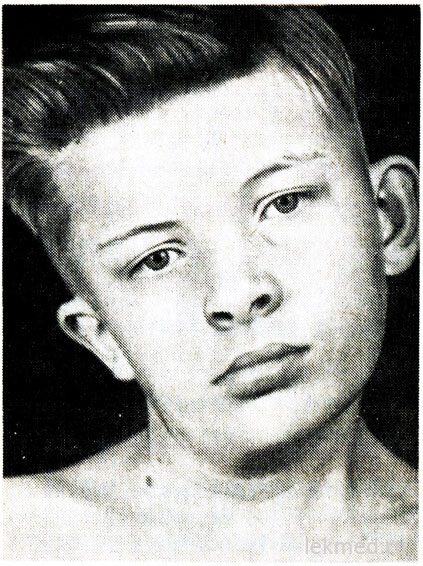
Vrodená torticollis je spôsobená jednostranným skrátením alebo kontraktúrou sternocleidomastoideus svalu. Hlava pacienta je naklonená ku kontraktúre a brada smeruje nadol v opačnom smere (obr. 21-7). Pri pokuse o korekciu polohy hlavy je cítiť výrazný svalový odpor. V postihnutom svale sú oblasti zhutnenia palpované. Príčina defektu je nejasná, dlho sa považovala za následok pôrodného poranenia. Torticollis sa však vyskytuje u detí narodených cisárskym rezom; to naznačuje, že v niektorých prípadoch sa príčina defektu týka vnútromaternicového obdobia. Torticollis treba odlíšiť od patologického záklonu hlavy v dôsledku deformácie krčných stavcov, ako je Klippel-Weilova anomália, a od zlomenín alebo dislokácií krčných stavcov. Röntgenovým vyšetrením sú vylúčené. U starších detí môže byť sklon hlavy spôsobený strabizmom, dystóniou, nádormi zadnej lebečnej jamky a krčnej miechy, myositis ossificans, krčnou lymfadenitídou alebo diafragmatickou herniou. Vo väčšine prípadov je možné vrodenú torticollis upraviť pomocou terapeutických cvičení. V chronickej forme však torticollis vedie k asymetrickému vývoju tváre a hlavy (pozri obr. 21-7), čo môže vyžadovať disekciu svalu, čo je spôsobené kozmetickými účelmi.
vrodené myopatie. Do tejto skupiny patrí niekoľko zriedkavých foriem dedičných ochorení, pri ktorých sa svalová slabosť a hypotónia objavujú už od dojčenského veku (pozri tabuľku 22-1). Ich presná diagnóza je veľký význam z hľadiska predpovede. Vo všeobecnosti je priaznivý pre normálnu životnú aktivitu a dĺžku života, na rozdiel od Werdnig-Hoffmannovej choroby alebo vrodenej svalovej dystrofie. Svalová biopsia zvyčajne pomáha identifikovať vrodené myopatie.
- Ochorenie centrálneho jadra. Centrálna časť svalových vlákien je sfarbená abnormálne, ale jednotne. Vyšetrenie elektrónovým mikroskopom odhalí pokles počtu mitochondrií a vyčerpanie sarkoplazmatického retikula v centrálnej časti vlákien.
Nemalínová myopatia. Pojem "nekarmínový" sa vysvetľuje skutočnosťou, že vo svalových vláknach sú určené vláknité štruktúry.
Ryža. 21-8. Myotonická kontrakcia jazyka (a) s prudkým úderom perkusného kladiva na jeho pravú polovicu a očné viečka (b) u dieťaťa s hyperkalemickou formou familiárnej periodickej paralýzy. 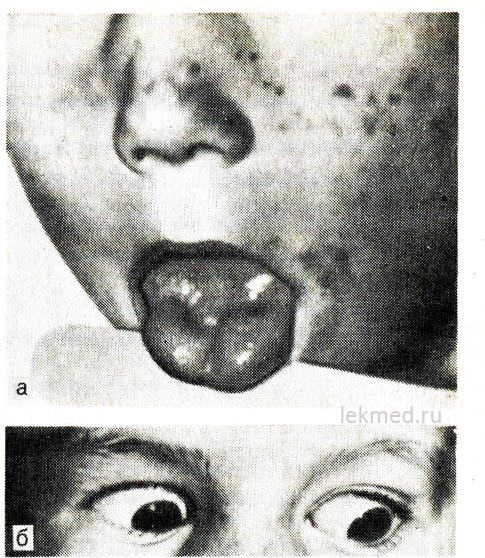
Pri pohľade nadol zostáva očné viečko stiahnuté.
Údaje z elektrónového mikroskopu naznačujú, že ide o výsledok zmien v Z-pásoch myofibríl.
Mitochondriálne myopatie. Boli hlásené niektoré formy myopatií, v ktorých najviac dôležité zmeny sa vyskytujú v mitochondriách svalových vlákien. Môžu sa výrazne zväčšiť ako v počte, tak vo veľkosti. Svalová slabosť a hypotónia môžu byť diagnostikované už v detstve, ale niekedy výrazne napredujú až v škole. Kardiomyopatia, encefalopatia a laktátová acidémia často sprevádzajú túto skupinu myopatií.
Myotónia. Tento stav je charakteristickým znakom rôznych svalových ochorení, ako je myotonia dystrophica, hyperkalemická familiárna paroxyzmálna obrna a ochorenia ukladania glykogénu. Myotónia je definovaná ako výrazné oneskorenie svalovej relaxácie po dobrovoľných alebo vynútených kontrakciách. Klinicky sa prejavuje neschopnosťou uvoľniť päsť alebo viditeľnou predĺženou kontrakciou svalov po ich stimulácii, ktorá sa prejavuje prudkým podráždením (obr. 21-8). Dá sa to pozorovať, ak perkusným kladivom zasiahnete povrchovú skupinu svalov, napríklad svaly jazyka alebo povrch dlane v oblasti elevácie prvého prsta. Myotónia je potvrdená elektromyografickými údajmi. V tomto prípade je charakteristická spontánna aktivita svalov badateľná po ich uvoľnení alebo dobrovoľnej kontrakcii (myotonické výboje).
Myotonia congenita (Thomsenova choroba). Jediným znakom tohto ochorenia, zdedeného dominantným typom, je myotónia. Môže sa prejaviť už v dojčenskom veku v podobe spomalenia prehĺtacích pohybov a následného zvracania
účinok neschopnosti normálnej relaxácie svalov hltanu. Vo vyššom detstve sa myotónia prejavuje ako neschopnosť pacienta uvoľniť prsty zovreté v päsť. Pri prvom pokuse o vykonanie nejakého pohybu svaly dieťaťa stvrdnú. Opakovaným opakovaním toho istého pohybu sa trochu uvoľnia. Takže napríklad choré dieťa zažíva veľké ťažkosti na začiatku aktu chôdze. Prvé kroky zvyčajne robí veľmi váhavo a pomaly. Po niekoľkých sekundách sa chôdza stáva normálnou alebo takmer normálnou. Symptómy myotónie sa zhoršujú nepriaznivým emočným stavom pacienta a ochladzovaním tela. Svalová sila zostáva v norme, svaly sú dostatočne vyvinuté a často nápadne zväčšené, čo vytvára mylný dojem o atletickej konštitúcii pacienta.
Diagnóza je založená na klinických nálezoch a elektromyografických údajoch. Aktivita sérových enzýmov je v rámci normálnych limitov. Jediným histologickým znakom je hypertrofia svalových vlákien.
Ochorenie sa líši od dystrofickej myotónie absenciou svalovej slabosti a atrofie a dystrofických zmien v biopsii svalového tkaniva. Liečba novokaínom alebo chinidínsulfátom je sprevádzaná účinkom a je indikovaná na funkčné poruchy. Priebeh ochorenia je zvyčajne benígny, s vekom sa môže stav pacienta zlepšovať.
paroxyzmálna paralýza. Táto skupina chorôb je charakterizovaná periodickou svalovou slabosťou s úplnou alebo takmer úplnou obnovou svalovej sily v období medzi záchvatmi. Zahŕňa tiež nedostatok svalovej fosforylázy (McArdleova choroba).
Hyperkalemická paroxyzmálna paralýza. Dedičná epizodická adynamia alebo paramyotónia sa prenáša podľa dominantného typu a je obzvlášť závažná u mužov. Zvyčajne začína v ranom detstve (niekedy v detstve). Útoky sa vyskytujú počas obdobia odpočinku po veľkom zaťažení svalov. Slabosť sa vyvíja rýchlo a môže trvať niekoľko hodín. Zvlášť sa to cíti v nohách; respiračná funkcia zvyčajne nie je narušená. Často je adynamia sprevádzaná myotóniou, ktorá pretrváva medzi záchvatmi, čo sa najzreteľnejšie prejavuje vo forme oneskorenia pohybu očných viečok pri pohľade nadol (pozri obr. 21-8, b).
Hladiny draslíka v sére sú často zvýšené počas záchvatu, ale na presné určenie môžu byť potrebné viaceré štúdie počas niekoľkých záchvatov. Útok je možné umelo vyvolať pomocou dávky draslíka (2-3 g perorálne), ale mal by sa vykonávať iba pod kontrolou EKG. Opakované útoky zastaví diakarb. Ťažké formy ochorenia sú charakterizované rozvojom chronickej, miernej slabosti a degeneratívnych zmien vo svaloch.
Hypokaliemická paroxyzmálna paralýza. Rodinná záchvatová paralýza, dedičná aj podľa dominantného typu, je obzvlášť náročná u chlapcov. Na rozdiel od hyperkalemickej formy sa prvý záchvat objavuje v neskorom detstve alebo v ranej adolescencii. Dôvodom je konzumácia bohatého jedla bohatého na sacharidy, prípadne oddych po cvičení. Zvyčajne útok začína nasledujúce ráno po ťažkej fyzickej námahe a výdatnej večeri. Je charakterizovaná svalovou slabosťou a areflexiou. Môže byť narušená funkcia dýchania. Môže sa pripojiť arytmia, vrátane ventrikulárneho extrasystolu a tachykardie. Záchvaty môžu trvať aj viac ako 24 hodín.V paralytickej fáze hladina draslíka v sére zvyčajne klesá (2-3 mmol/l). Základná chyba nie je známa. U pacientov s opakovanými ťažkými záchvatmi sa rozvinie chronická svalová slabosť a patologické zmeny vo svaloch. Liečba počas záchvatov spočíva v užívaní chloridu draselného; jeho počiatočná dávka je 2-3 g Diakarb pomáha znižovať frekvenciu záchvatov.
Paroxyzmálna myoglobinúria (idiopatická myoglobinúria). Idiopatická myoglobinúria je heterogénna skupina porúch, pri ktorých sa záchvaty paralýzy s myoglobinúriou vyskytujú spontánne alebo po intenzívnom cvičení. Choroba sa dedí dominantným spôsobom, spojený s chromozómom X. Svaly, najčastejšie lýtka a stehná, sú pri záchvate bolestivé a opuchnuté. Moč sa stáva tmavočervenou alebo hnedou. Myoglobinúria môže spôsobiť renálnu tubulárnu nekrózu, ktorá vedie k smrti v dôsledku zlyhania obličiek.
Diagnóza je potvrdená detekciou myoglobulínu v moči. Pozitívny benzidínový test v neprítomnosti erytrocytov v moči potvrdzuje prítomnosť myoglobínu v ňom, najmä ak hemoglobín nie je zistený v sére. Hemoglobín sa stanovuje spektrofotometriou. Paroxyzmálnu myoglobinúriu treba odlíšiť od McArdleovej choroby, deficitu karnitín palmityltransferázy a myoglobinúrie po nezvyčajnom namáhavom cvičení alebo svalovom poranení u zdravého jedinca. Myoglobinúria po ťažkom svalovom cvičení sa vyskytuje pri pseudohypertrofickej svalovej dystrofii (Duchennova choroba).
Liečba pozostáva z odpočinku na lôžku; ak je to potrebné, vykonajte umelú ventiláciu pľúc. Aby sa zabránilo zlyhaniu obličiek, je potrebné pacientovi predpísať bohatý nápoj.
Nedostatok karnitín palmityltransferázy. Pri nedostatku tohto enzýmu je narušený prenos mastných kyselín s dlhým reťazcom do mitochondriálnych segmentov, v ktorých prebieha oxidácia a tvorba ketónov. Nedostatok izoenzýmu typu II sa dedí recesívnym spôsobom. V dôsledku jeho nedostatku je narušená ketogenéza v tkanivách vrátane svalov a pečene. Prvé príznaky ochorenia sa objavujú častejšie u detí v škole a dospievaní. Pozostávajú z opakovaných epizód svalovej bolesti, slabosti a horúčky po cvičení alebo nalačno. Myoglobinúria sprevádzajúca záchvaty môže viesť k zlyhaniu obličiek. Pôst vedie k hypoglykémii. Medzi útokmi sa deti javia ako zdravé. Ochorenie sa musí odlíšiť od iných stavov sprevádzaných periodickou slabosťou a myoglobinúriou. Metóda stanovenia aktivity karnitín palmityl transferázy má diferenciálnu diagnostickú hodnotu. Klesá vo svalových a pečeňových tkanivách, leukocytoch a fibroblastovej kultúre. Konzumácia nízkotučnej stravy bohatej na sacharidy môže pomôcť znížiť záchvaty.
Svalové dystrofie. Tieto anomálie patria do skupiny rodinných ochorení sprevádzaných degeneráciou svalových vlákien. Klasifikácia svalových dystrofií je založená na znakoch, ako je čas nástupu, rýchlosť progresie, distribúcia lézií podľa svalovej skupiny a spôsob dedičnosti.
Pseudohypertrofická svalová dystrofia. Detstvo alebo Duchennova forma je najbežnejšou formou svalovej dystrofie; jeho frekvencia je 0,14 na 1000 detí. V klasickej forme sa vyskytuje len u chlapcov a dedičnosť viazaná na X chromozóm sa vyskytuje približne u 50 % probandov. V iných prípadoch je choroba spôsobená novými mutáciami. Uvádza sa zriedkavá forma svalovej dystrofie, klinicky identická s Duchennovou formou, ale dedičná recesívnym typom s rovnakou frekvenciou ochorenia u chlapcov a dievčat. Spoľahlivo diagnostikovať ochorenie je zriedka možné u dieťaťa mladšieho ako 3 roky. V anamnéze sú väčšinou náznaky, že dieťa malo spomalený vývin pohybových funkcií, začalo neskoro sedieť, chodiť, behať, čo prirodzene svedčí o skoršom nástupe ochorenia. Častými klinickými prejavmi sú kolísavá (kačacia) chôdza, ťažkosti pri chôdzi po schodoch a hypertrofia lýtkových svalov. V niektorých prípadoch sú do procesu zapojené aj iné svaly, najmä deltový sval, brachioradialis a svaly jazyka.
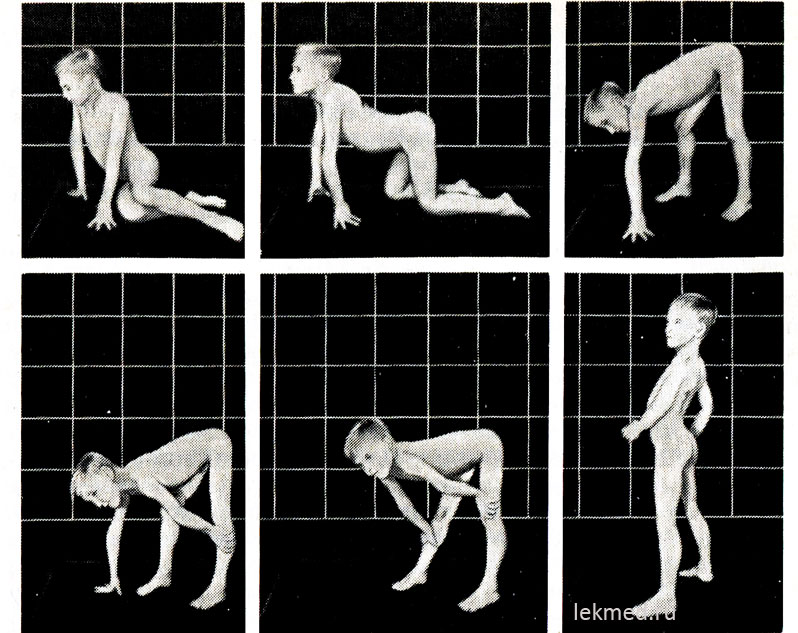
Ryža. 21-9. Typické polohy zaujaté pri vstávaní z podlahy (znak Govers) u 7-ročného dieťaťa s pseudohypertrofickou myopatiou.
V stoji (posledná fotografia) je výrazne výrazná lordóza.
Na začiatku ochorenia majú hypertrofované svaly výraznú silu, ale neskôr sa znižuje (pseudohypertrofia), pretože k nárastu svalovej hmoty dochádza v dôsledku ich tukovej infiltrácie. Sila hypertrofovaného svalu gastrocnemia výrazne prevyšuje silu svalov prednej plochy nohy, čo vysvetľuje časté kontraktúry pätovej šľachy a chod dieťaťa po prstoch. Slabosť svalov panvového pletenca sa prejavuje charakteristickou kačacou (pánovou) chôdzou a ťažkosťami, ktoré dieťa zažíva, keď vstáva zo sedu na podlahe. Pri dostatočne ťažkých formách svalovej dystrofie má dieťa Goversov príznak: vstáva z podlahy, najprv si kľakne, opiera sa o ruky a potom sa zdvihne, pričom postupne odtláča ruky od holene, kolenných kĺbov a bedier (obr. 21-9). Slabosť svalov ramenného pletenca zistíte tak, že dieťa držíte vo zvýšenej polohe za podpazušie. Normálne sa snaží udržať pritlačením rúk k telu; pri svalovej dystrofii akoby prekĺzla cez ruky skúšajúceho. Choré dieťa často nedokáže zdvihnúť ruky nad hlavu. V neskorších štádiách ochorenia vzniká výrazná svalová atrofia. Zvyčajne vo veku 12 rokov už dieťa nemôže chodiť. Pacienti v 75 % prípadov zomierajú pred dosiahnutím veku 20 rokov. Väčšina z nich má kardiomyopatiu, ktorá v niektorých prípadoch spôsobuje náhlu smrť. Ak je dedičnosť viazaná na X a choroba sa začala v staršom detstve, očakávaná dĺžka života zostáva dlhá (Beckerova svalová dystrofia). Priemerné IQ detí s DMD je 80; 25% detí má mentálnu retardáciu.
Pri diferenciálnej diagnostike Duchennovej svalovej dystrofie je potrebné mať na pamäti Werdnigovu-Hoffmannovu chorobu u starších dojčiat a svalové choroby, ako sú endokrinné myopatie, nedostatok karnitínu, choroby ukladania glykogénu a polymyozitída. Niekedy pri kontraktúrach kalkaneálnej šľachy a chôdzi dieťaťa po prstoch na nohách možno predpokladať detskú mozgovú obrnu, ale pri svalovej dystrofii nie sú spasticita a hyperreflexia charakteristické pre detskú mozgovú obrnu.
Diagnóza je založená na stanovení aktivity enzýmov v sére, elektromyografických údajoch a biopsii svalového tkaniva. Aktivita enzýmov, najmä kreatínfosfokinázy, často prekračuje normu 10-krát aj u dojčiat ešte pred rozvojom klinických príznakov. Na elektromyograme sa najprv odhalí skrátenie trvania a zníženie amplitúdy motorických potenciálov. Histologické zmeny pozostávajú z degenerácie svalových vlákien. Často sa líšia veľkosťou a sú čiastočne nahradené tukom a spojivovým tkanivom. Veľkosť ich jadier sa tiež líši. Diagnózu možno stanoviť pri narodení stanovením aktivity kreatínfosfokinázy. Metódy identifikácie prenášačiek ešte neboli vyvinuté, napriek tomu, že 60 – 80 % z nich vykazuje mierny alebo mierny nárast jeho hladiny. Tieto znaky sú typické skôr pre detstvo ako pre nasledujúce obdobia života.
Neexistujú žiadne účinné liečby. Pacient by mal byť aktívny a mal by byť schopný čo najviac chodiť. Je potrebné zabezpečiť, aby sa dieťa vyhýbalo intenzívnej fyzickej aktivite, pretože môže spôsobiť pretrhnutie svalových vlákien. V niektorých prípadoch chirurgické predĺženie kalkaneálnej šľachy zlepšuje schopnosť chôdze, ale predĺžený odpočinok na lôžku po ortopedickej korekcii môže zhoršiť svalovú atrofiu. Genetické poradenstvo zohráva dôležitú úlohu.
Vrodená svalová dystrofia. Ochorenie sa dedí autozomálne recesívnym spôsobom a je charakterizované svalovou hypotenziou a slabosťou u dojčiat. Je zaradený do skupiny stavov definovaných ako „lenivé dieťa“ (pozri tabuľku 21-1). Nástup ochorenia sa vzťahuje na vnútromaternicové obdobie. Niekedy má novorodenec výraznú atrofiu svalov, ich kontraktúry, obmedzenú pohyblivosť kĺbov. Odlíšenie od Werdnig-Hoffmannovej choroby je ťažké. Fascikulácie jazyka, charakteristické pre druhý, chýbajú pri svalovej dystrofii. Šľachové reflexy sú potlačené, ale nie úplne stratené. Tento proces zahŕňa svaly zapojené do dýchania, vrátane bránice. V závažných prípadoch nastáva smrť pred dosiahnutím veku 1 roka v dôsledku zlyhania dýchania; pri ľahších formách sa dlhodobo udržiava normálna životaschopnosť. Zvýšenie aktivity sérových enzýmov nie je zaznamenané, hoci vo svaloch sa vyskytujú dystrofické zmeny.
Rameno-tvárová forma svalovej dystrofie. Táto pomerne mierna forma svalovej dystrofie sa dedí autozomálne dominantným spôsobom. Zvyčajne začína vo veku 10-20 rokov a je charakterizovaná slabosťou a atrofiou svalov tváre a ramenného pletenca. Tvár je úplne amimická, pacient nemôže zavrieť oči a vydať píšťalku. Choroba postupuje pomaly a je kompatibilná s normálnou dĺžkou života. Diagnóza je založená na klinickom náleze a type dedičnosti. Výsledky biopsie svalového tkaniva naznačujú dystrofické zmeny v ňom. Hladiny kreatínfosfokinázy v sére môžu zostať normálne alebo mierne zvýšené.
Panvová forma svalovej dystrofie. Táto skupina heterogénnych porúch je charakterizovaná pomalou progresiou svalovej dystrofie a je dedená autozomálne recesívnym spôsobom. Začiatok ochorenia sa vzťahuje na staršie detstvo, dospievanie alebo dospelosť. Zvyčajne sú postihnuté svaly panvového pletenca.
Očná forma myopatie. Dystrofické zmeny sa vyskytujú najmä vo vonkajších očných svaloch. Choroba začína v detstve alebo dospievaní. S ním postupuje ptóza a obmedzenie pohybov očnej gule. Niekedy sa slabosť rozširuje na svaly tváre a krku. Ochorenie treba odlíšiť od myasthenia gravis a obrny hlavových nervov pri nádoroch mozgového kmeňa.
Progresívna oftalmoplégia, začínajúca v detstve alebo dospievaní, je spojená s atypickou retinitis pigmentosa a srdcovým blokom (Kearns-Sayersov syndróm). Zvyčajne sa spája s progresívnou ataxiou, oneskoreným rastom a pubertou. Pod sarkolemou svalov sa určujú veľké akumulácie atypických mitochondrií. Genetická povaha tohto procesu nebola stanovená. Možnosť náhlej smrti v dôsledku porušenia srdcového vedenia môžete kontrolovať pomocou kardiostimulátora.
Myotonická dystrofia. Napriek tomu, že myotonická dystrofia začína akoby u dospelého človeka, jej nástup čoraz častejšie zaznamenávame už u dojčiat a neskôr u detí. Dedí sa autozomálne dominantným spôsobom. Jeho nástup v detstve naznačuje, že matka trpí myotóniou. V súlade s tým môžu vnútromaternicové faktory ovplyvniť závažnosť ochorenia u dieťaťa. Už pri narodení môže mať svalovú hypotenziu, chýba mu sať. Zaostávanie fyzického a duševného vývoja sa zvyčajne zistí neskôr. V ranom detstve sa svalová slabosť a atrofia rozšírili hlavne na tvárové, čeľustné a temporálne svaly. Zvyčajne sa zaznamenáva bilaterálna ptóza. Medzi diagnosticky významné metódy patrí svalová perkusia, elektromyografia; pre týchto pacientov je typická neschopnosť uvoľniť ruku zovretú v päsť (pozri Vrodená myotónia). Slabosť a atrofia svalov končatín a panvového pletenca (zvyčajne distálne skupiny) sa zisťujú vo vyššom detstve alebo dospievaní. U dospelých je toto ochorenie sprevádzané šedým zákalom, plešatosťou, atrofiou semenníkov.
Diagnóza je založená na identifikácii príznakov myotónie, charakteristického rozloženia svalovej slabosti, dedičnosti podľa dominantného typu a dystrofických zmien vo svaloch. V detskom veku môže byť priebeh ochorenia nepriaznivý, často sprevádzaný mentálna retardácia. V období dospievania sa do popredia dostáva svalová slabosť. Pri funkčných poruchách je indikovaná liečba novokaínom a chinidínom.













