Diagramele de echilibru de fază sunt baza științei materialelor. Diagrame de fază ale sistemelor bicomponente „polimer-solvent” Diagrame de fază ale sistemelor bicomponente
Diagrame de fază ale sistemelor cu două componente "polimer - solvent"
Este descris comportamentul de fază al soluțiilor adevărate regula fazei Gibbs. Pentru sistemele incompresibile condensate, când influența presiunii poate fi neglijată, regula fazei se scrie ca
unde / este numărul de grade de libertate, adică numărul de variabile termodinamice independente (temperatura, concentrația etc.) care pot fi modificate în mod arbitrar fără modificarea numărului de faze din sistem și fără încălcarea echilibrului acestuia; LA - numărul de componente; Ф - numărul de faze.
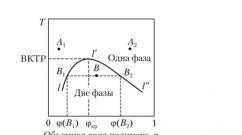
O reprezentare grafică a regulii fazei Gibbs este o diagramă de stări sau diagramă de fază, care pentru sistemele cu două componente „polimer amorf - lichid cu greutate moleculară mică” (K = 2) are forma unei curbe de solubilitate sau a unei curbe de amestecare reciprocă a componentelor în coordonatele „temperatura – compoziție”. Pe fig. 2.3 prezintă un exemplu de astfel de diagramă de fază.

Orez. 2.3.
"polimer - solvent"
Compoziția este de obicei exprimată în greutate (w) sau fracții de volum (av) ale polimerului. Zona de deasupra curbei corespunde unui sistem monofazat. De exemplu, la puncte A j sau L 2:/ = 2 - 1 + 1 = 2, adică. Sistemul are două grade de libertate. Aceasta înseamnă că temperatura și compoziția pot fi modificate simultan fără a modifica numărul de faze din sistem. În acest caz, punctul L, corespunde adevăratei soluții a polimerului în solvent și punctul A 2- o soluție adevărată a unui lichid cu greutate moleculară mică într-un polimer.
Zona de sub curbă corespunde unui sistem în două faze. Da, la punct ÎN sistemul este stratificat în două faze: compoziţie q>(B x)și compoziția f (LA 2). Faza d este îmbogățită în solvent (soluție a polimerului într-un solvent) și faza LA 2- component polimeric (soluție dintr-un polimer dintr-un component cu greutate moleculară mică). Linia a, - ÎN- d 2 , paralel cu axa x și care leagă fazele care coexistă în echilibru, se numește nodul(din lat. nodus- nod, unire, centură). Raportul volumelor celor două faze este invers proporțional cu raportul valorilor segmentelor dintre puncte B contra Bși d 2 („regula pârghiei”), adică

Linia 1-Y-Y, separând intervalele de compoziții corespunzătoare sistemelor monofazate și bifazate, se numește bipodalio. Pentru fiecare temperatură din interiorul regiunii de separare a fazelor este satisfăcută egalitatea / = 2-2 + 1 = 1. Aceasta înseamnă că în interiorul regiunii delimitate de linia binodă, sistemul are un grad de libertate, adică. o variabilă (temperatura sau concentrația de fază) determină complet starea sistemului. Fiecare temperatură are propria sa compoziție de faze coexistente și invers. În acest caz, proprietățile sistemului în fiecare punct al diagramei de fază nu depind de modul de atingere a echilibrului: diluare, concentrare, răcire sau încălzire. Dacă moleculele componentelor sunt comparabile ca mărime, atunci diagrama de fază este simetrică în raport cu axa compozițiilor. Curbele de amestecare polimer-solvent sunt întotdeauna puternic asimetrice: sunt deplasate în mod vizibil către concentrații scăzute de polimer.
Temperatura critică superioară de dizolvare (UCTR) este temperatura minimă peste care nu se observă nicio separare a soluției de polimer la nicio concentrație a soluției de polimer.
Se numește concentrația soluției de polimer corespunzătoare VCTR concentrare critică(A se vedea figura 2.3).
Sistemele cu VCTR sunt caracteristice cazului de dizolvare endotermă a polimerului, când căldura este absorbită în timpul dizolvării, c.t.
unde AR cm este modificarea entalpiei în timpul dizolvării; # R. 1STI)p este entalpia soluției; Eu si N., sunt entalpiile solventului pur și respectiv polimerului. Aici și mai jos, indicele 1 se referă la solvent, indicele 2 la polimer.
Formarea spontană a soluțiilor adevărate este însoțită de o scădere a potențialului izobar-izotermic al sistemului:
Deoarece DS cm \u003d DYA CH - 7D5 SM (unde D5 SM \u003d 5 soluție - 5, - 5 2 este modificarea entropiei amestecării în timpul dizolvării), atunci D5 C\u003e | > 0 (modificarea entropiei este pozitivă), iar DYa vezi TAS m . Ultima inegalitate înseamnă că dizolvarea spontană a polimerului este posibilă numai la temperaturi peste o anumită valoare critică T> AN/AS. Exemple tipice de sisteme cu VCTR sunt sistemele „polimer cu polaritate scăzută - solvent cu polaritate scăzută”, de exemplu, „polistiren - ciclohexan”, „poliizobutilenă - benzen”, „acetat de celuloză - cloroform”, etc.
Există sisteme care sunt temperatură critică de dizolvare mai scăzută(Fig. 2.4).
Temperatura critică de dizolvare inferioară (LCST) este temperatura maximă sub care nu se observă nicio separare a soluției de polimer la nicio concentrație.
Sistemele cu LCST sunt tipice pentru cazul dizolvării exoterme a polimerului, când căldura este eliberată în timpul dizolvării (DYa cm T Acest lucru se observă, de exemplu, la dizolvarea polimerilor polari în solvenți polari.
Modificarea entalpiei este negativă datorită efectelor energetice „puternice” asociate cu formarea legăturilor de hidrogen sau acumulării de donatori.

Orez. 2.4. Un exemplu de diagramă de fază cu LCST pentru un sistem cu două componente „polimer-solvent” a interacțiunilor primare dintre unitățile polimerice și moleculele de solvent. În același timp, entropia în timpul dizolvării scade și datorită imobilizării solventului în învelișurile de solvat ale macromoleculelor.
Din punct de vedere fizic, motivele apariției LCST sunt asociate, în primul rând, cu distrugerea interacțiunilor specifice menționate mai sus dintre polimer și solvent la încălzire, ceea ce înrăutățește compatibilitatea componentelor. În al doilea rând, aspectul LCST este determinat de diferența dintre coeficienții de dilatare termică volumetrică a polimerului și solventului. Când este încălzit, solventul se extinde mai puternic, comprimând macromoleculele și facilitând formarea asociaților cu eliberarea lor ulterioară într-o nouă fază.
Exemple de sisteme cu LCST sunt sistemele „iolietilen oxid – apă”, „metilceluloză – apă”, „nitroceluloză – alcool etilic”, etc.
Atunci când DY SM și D5 (. m semnează invers cu o schimbare a temperaturii (de exemplu, datorită reorganizării structurii soluției din cauza unei modificări a naturii interacțiunii moleculelor de solvent și a segmentelor de polimer), un comportament de fază mai complex În acest caz, diagramele de fază sunt combinații ale diagramelor de mai sus cu VCTR și LCTR Două variante ale unor astfel de diagrame de fază sunt prezentate în Fig. 2.5.

Orez. 2.5.
A- NCTR 6 - NCTR > VKTR
Diagrama de fază sub forma unei bucle închise (Fig. 2.5, A), caracteristic unei soluții de oxid de polipropilenă în apă. Diagrame de fază de al doilea tip (Fig. 2.5, b) se observă pentru sistemele „polistiren – ciclohexan”, „polietilenă – alcani”, „poliacetat de vinil – metanol”, „alcool polivinilic – apă”, „poliizobutilenă – benzen” etc.
REOLOGIE- știința deformărilor și fluidității mediilor continue care prezintă proprietăți elastice, plastice și vâscoase în diferite combinații. Deformarile elastice apar in corp atunci cand se aplica o sarcina si dispar daca sarcina este indepartata; deformarile plastice apar atunci cand tensiunile cauzate de sarcina depasesc o valoare cunoscuta - limita de curgere; persistă după descărcare; curgerea vâscoasă se distinge prin faptul că are loc la orice tensiuni arbitrar mici, odată cu creșterea tensiunilor, viteza curgerii crește și, menținând tensiunile, curgerea vâscoasă continuă la nesfârșit. O altă proprietate care poate fi deținută de mediile studiate de reologie este elasticitatea ridicată, care este tipică, de exemplu, pentru cauciuc, atunci când o bandă de cauciuc permite întinderea de zece ori, iar după îndepărtarea încărcăturii, aproape instantaneu își restabilește starea inițială.
Un proces reologic tipic este o curgere relativ lentă a unei substanțe în care se găsesc proprietăți elastice, plastice sau foarte elastice. Fenomenele reologice se manifestă în multe procese naturale și într-un număr mare de procese tehnologice. Substanțele implicate în astfel de procese sunt foarte numeroase: acestea sunt rocile care alcătuiesc scoarța terestră, magma, lava vulcanică, acestea sunt soluții de petrol și argilă care joacă un rol crucial în producția de petrol; argilă umedă, pastă de ciment, beton și beton asfaltic (un amestec de asfalt și nisip care acoperă trotuarul), acestea sunt vopsele în ulei - un amestec de particule de ulei și pigment; acestea sunt soluții și topituri de polimeri în procesul de fabricare a firelor, foliilor, țevilor prin extrudare; in sfarsit, acestea sunt aluatul de paine si masele pastoase, din care se fac dulciuri, carnati, creme, unguente, paste de dinti, acesta este combustibil solid pentru rachete; acestea sunt, în sfârșit, corpuri proteice, de exemplu, țesuturile musculare.
TERMODINAMICA DULUI SPONTAN A POLIMERILOR
Dizolvarea polimerilor este similară cu amestecarea nelimitată a două lichide, care respectă cea de-a doua lege a termodinamicii. A doua lege a termodinamicii este un model general care vă permite să găsiți direcția și să stabiliți posibilitatea sau imposibilitatea proceselor termodinamice. Conform celei de-a doua legi, căldura nu se poate transfera spontan de la un corp mai puțin cald la unul mai cald.
Procesele în care sunt implicate fenomenele termice merg spontan într-o singură direcție și se opresc după ce se ajunge la starea de echilibru termodinamic - o stare mai „preferată”. Entropia este o funcție de stare care caracterizează măsura acestei „preferințe”. Într-un sistem izolat, sunt posibile doar astfel de procese în care entropia crește sau rămâne neschimbată.
Condiția termodinamică pentru dizolvarea spontană - modificarea energiei libere a sistemului în timpul dizolvării spontane trebuie să fie negativă:
ΔG = ΔH-TΔS<0,
unde ΔН este modificarea entalpiei sau entalpia amestecării; ΔS este modificarea entropiei sau entropia amestecării; T este temperatura absolută. Există trei cazuri posibile aici:
1) ΔG<0- при растворении происходит поглощение тепла (эндотермическое растворение);
2) ΔG>0 - în timpul dizolvării se eliberează căldură (dizolvare exotermă);
3) ΔG=0 - nicio căldură nu este absorbită sau eliberată (dizolvare atermică).
Respectarea condiției de dizolvare termodinamică ΔG<0 возможно при следующих условиях:
1) în condiția Δ H< 0, которое соблюдается, если при растворении выделяется теплота, так как изменение энтальпии (или внутренней энергии) равно интегральной теплоте растворения с обратным знаком. Такое условие часто соблюдается на практике, например, при растворении полярных полимеров в полярных растворителях. Положительный тепловой эффект при растворении объясняется тем, что теплота сольватации макромолекул больше теплоты собственно растворения, а как известно, общий тепловой эффект растворения равен алгебраической сумме теплот сольватации и собственно растворения;2) în condiția Δ S> 0, care se realizează întotdeauna în practică în timpul dizolvării, deoarece entropia amestecării este întotdeauna pozitivă. Entropia amestecării HMS cu un solvent, calculată pe fracția de greutate a unei substanțe, se află între valorile entropiei de dizolvare a substanțelor cu greutate moleculară mică și sistemele coloidale tipice.
Adesea, atunci când DIU este dizolvat, procesul de dizolvare are loc exclusiv datorită unei modificări a entropiei (în direcția creșterii), adică termic. De asemenea, trebuie luat în considerare efectul temperaturii. Dacă polimerul nu se dizolvă la o anumită temperatură (ΔН-TΔS>0), atunci odată cu creșterea temperaturii, valoarea absolută a TΔS poate deveni mai mare decât valoarea absolută a ΔН, iar apoi semnul de inegalitate se va schimba la opus. Această temperatură se numește temperatura critică de amestecare.
Teoria soluțiilor de polimeri, dezvoltată folosind termodinamică și bazată pe analogia dintre dizolvarea polimerilor și amestecarea nerestrânsă a două lichide, are o serie de neajunsuri din cauza numeroaselor ipoteze și corecții în determinarea atât a entropiei amestecării ΔS cât și a căldurii. de amestecare ΔН a unui polimer cu un solvent.
Echilibrul de fază în sistemul polimer-solvent
Stabilitatea sistemului (echilibrul de fază), de regulă, este determinată de gradul de afinitate termodinamică a componentelor, depinde de compoziția și structura chimică a acestora, de condițiile externe, în special de temperatură. Legea principală de echilibru a unui sistem multifazic multicomponent este regula fazelor Gibbs, care stabilește relația dintre numărul de faze F, numărul de componente din sistemul K și numărul gradelor sale de libertate C:
Numărul de grade de libertate arată câte variabile termodinamice care determină starea sistemului (presiune, temperatură etc.) pot fi modificate în mod arbitrar fără a provoca o modificare a numărului de faze din sistem, adică fără a perturba echilibrul acestuia. .
În sistemele în care componentele sunt doar în stare lichidă și solidă, modificarea presiunii are un efect redus asupra proprietăților, astfel încât presiunea poate fi considerată constantă, iar ecuația regulii fazei ia forma
Conform acestei ecuații, un sistem monofazat cu două componente are două grade de libertate (starea sistemului este determinată de temperatura și concentrația unuia dintre componente). În prezența a două faze (Ф = 2), sistemul cu două componente are un grad de libertate. Aceasta înseamnă că o modificare a temperaturii determină o modificare a concentrației ambelor faze. La o anumită temperatură, aceste faze se pot îmbina pentru a forma o soluție omogenă monofazată. În schimb, o soluție omogenă monofazată la o anumită temperatură se poate delamina sau se poate separa în două faze. Temperatura la care are loc separarea se numește separare de fază sau temperatură de separare a fazelor (TfP). O soluție a fiecărei concentrații are propriul GphR, a cărui dependență de compoziția soluției este exprimată printr-o curbă de amestecare reciprocă sau o curbă limită care separă regiunea soluțiilor monofazate de cele bifazate.
În funcție de compoziția soluțiilor lichide monofazate bicomponente în timpul răcirii lor, sunt posibile două cazuri de separare în componente constitutive: lichid și cristalin. Cu separarea lichidă, o fază lichidă este separată în două faze lichide, cu separarea cristalină, o componentă sub formă de fază cristalină se separă de soluție.
Orez. 10.6. Diagrame de echilibru de fază a sistemelor HMS - solvent: a) sistem cu VCTR; b) sistem cu LCTR; c) sistem cu VKTR și LCTR; d) sistem cu VKTR Orez. 10.6. Diagrame de echilibru de fază a sistemelor HMS - solvent: a) sistem cu VCTR; b) sistem cu LCTR; c) sistem cu VKTR și LCTR; d) sistem cu VKTR Orez. 10.6. Diagrame de echilibru de fază a sistemelor HMS - solvent: a) sistem cu VCTR; b) sistem cu LCTR; c) sistem cu VKTR și LCTR; d) sistem cu VKTR Orez. 10.6. Diagrame de echilibru de fază a sistemelor HMS - solvent: a) sistem cu VCTR; b) sistem cu LCTR; c) sistem cu VKTR și LCTR; d) sistem cu VKTR Orez. 10.6. Diagrame de echilibru de fază a sistemelor HMS - solvent: a) sistem cu VCTR; b) sistem cu LCTR; c) sistem cu VKTR și LCTR; d) sistem cu VKTR Orez. 10.6. Diagrame de echilibru de fază a sistemelor HMS - solvent: a) sistem cu VCTR; b) sistem cu LCTR; c) sistem cu VKTR și LCTR; d) sistem cu VKTR Orez. 10.6. Diagrame de echilibru de fază a sistemelor HMS - solvent: a) sistem cu VCTR; b) sistem cu LCTR; c) sistem cu VKTR și LCTR; d) sistem cu VKTR Orez. 10.10. Modificarea celui de-al doilea coeficient virial cu temperatura: 1- polistiren - ciclohexan; 2 - poliizobutilenă - n-pentan; 3 - poliizobutilenă - dibutil eter
Procesele de interacțiune a polimerilor cu lichide cu greutate moleculară mică sunt de mare importanță în sinteza polimerilor, prelucrarea lor în produse și în condițiile de funcționare a acestor produse în diferite medii lichide. Procesul de plastificare este legat și de așa-numita compatibilitate a plastifianților cu polimeri, adică. cu formarea de soluţii adevărate. În cele din urmă, soluțiile de polimeri sunt de mare importanță pentru determinarea greutăților moleculare ale polimerilor și a dimensiunilor macromoleculelor.
Când polimerul interacționează cu lichide cu greutate moleculară mică, soluții adevărate, sisteme coloidaleși studenți. Următoarele sunt semne ale unei soluții adevărate și ale unui sistem coloidal:
Document fără nume
Soluții adevărate |
sisteme coloide |
| 1. Prezența afinității între componente | 1. Lipsa de afinitate între componente |
| 2. Educație spontană | 2. Educație nespontană |
| 3. Dispersie moleculară sau ionică | 3. Dispersia coloidală |
| 4. Stabilitate termodinamică | 4. Instabilitate termodinamică |
| 5. Cresterea gradului de dispersie | 5. Reducerea gradului de dispersie în timp |
| 6. Stabilitate agregativă | 6. Instabilitate agregativă |
| 7. Monofazat | 7. Bifazic |
| 8. Fără interfețe | 8. Prezența interfețelor |
| 9. Reversibilitate | 9. Ireversibilitate |
Toate aceste semne sunt interconectate. Într-adevăr, dacă există o afinitate între componente, atunci la contactul direct unul cu celălalt, fără nicio cheltuială de energie externă, ele încep să se disperseze spontan unul în celălalt, ceea ce duce la o creștere treptată a gradului de fragmentare sau a gradului de fragmentare. dispersie în moleculară sau ionică.
Dispersia spontană sau dizolvarea, ca orice proces spontan care are loc la presiune și temperatură constantă, este însoțită de o scădere a energiei libere Gibbs (), care este tipică pentru un sistem stabil termodinamic. În acest caz, se formează un sistem monofazat în care nu există interfețe.
În orice soluție adevărată, dacă nu este diluată la infinit, ca urmare a interacțiunii moleculelor de dizolvat între ele, se formează asociații care sunt distruși reversibil sub influența mișcării termice. Acest lucru duce la posibilitatea unor modificări reversibile ale proprietăților soluției atunci când condițiile externe se modifică. Astfel, o soluție adevărată din două componente poate fi încălzită; se răcește, se diluează, se concentrează, dar la o anumită temperatură și presiune, concentrația soluției, proprietățile și structura acesteia vor fi aceleași, indiferent de modul în care este preparată soluția. Un echilibru care nu depinde de calea pentru a-l atinge se numește adevărat. De aici chiar numele soluțiilor.
Dacă nu există afinitate între componente, atunci oricât de mult sunt în contact, dispersia spontană nu are loc. (O bucată de aur nu va începe să se dizolve în apă.) Pentru a zdrobi componenta sunt utilizate diferite tipuri de energie, de exemplu, energia mecanică, care este transformată în energie liberă a sistemului (exemplu "> latex. Aceasta este o dispersie apoasă sau o emulsie de picături de cauciuc în apă. Astfel de dispersii de cauciucurile sintetice se numesc latexuri sintetice.
Pentru a preveni coalescerea picăturilor în emulsie și pentru a face sistemul stabil, este necesar un stabilizator. În latexul natural, stabilizatorii sunt diferite substanțe proteice, ale căror molecule sunt adsorbite la interfața picătură-mediu. În latexurile sintetice, stabilizatorul este un emulgator ale cărui molecule sunt, de asemenea, adsorbite pe interfața polimer-mediu. Pentru latexuri sunt valabile toate regularitățile caracteristice sistemelor coloidale.
Soluțiile de compuși cu molecul mare, precum și substanțele cu molecul scăzut, pot fi împărțite în soluții non-electrolitice și soluții electrolitice. Soluțiile de neelectroliți formează polimeri care nu se disociază în ioni, iar soluțiile de electroliți formează polimeri, la dizolvarea cărora are loc disocierea electrolitică.
Primit în prezent polielectroliți sintetici- polimeri capabili să se disocieze în ioni în soluții, iar într-o macromoleculă apar un număr mare de sarcini care se repetă periodic. Exemple de poliacizi sunt poliacrilici și acizi polimetacrilici. Sărurile solubile în apă ale acestor acizi se disociază în soluție:
exemplu">polibază este polivinilpiridiniu:
exemplu"> polielectroliți reticulați, care se obțin prin introducerea grupărilor ionizante în diverși polimeri de rețea.
Soluțiile adevărate de polimeri formate spontan au toate caracteristicile de mai sus ale soluțiilor adevărate. Dar, din cauza diferenței uriașe de dimensiune a moleculelor de polimer și solvent, soluțiile polimerice adevărate au propriile caracteristici specifice, care includ fenomenul de umflare și vâscozitatea ridicată a soluțiilor chiar diluate.
Dacă turnați cu atenție un lichid peste un strat al altuia, are loc pătrunderea lor reciprocă; în cazul apei și alcoolului etilic, moleculele acestuia din urmă pătrund în faza apoasă, iar moleculele de apă în faza alcoolică. Deoarece moleculele ambelor lichide sunt mici și mobile, pătrunderea lor reciprocă are loc cu aceeași viteză, iar lichidele se amestecă.
Particularitatea dizolvării polimerului este că componentele sunt amestecate, ale căror dimensiuni ale moleculelor diferă de mii de ori; de unde mobilitatea diferită a moleculelor. Mobilitatea moleculelor unui lichid cu greutate moleculară mică este foarte mare. Când un polimer intră în contact cu un lichid cu greutate moleculară mică, moleculele sale încep să pătrundă rapid în faza de polimer, iar macromoleculele uriașe nu au timp să treacă în faza de solvent în acest timp: înainte de dizolvare, polimerul cu greutate moleculară mare se umflă. .
Umflarea este procesul de absorbție sau sorbție a lichidelor cu greutate moleculară mică (sau a vaporilor acestora) de către un polimer.
Dar acest proces este fundamental diferit de procesele de adsorbție fizică care au loc pe suprafața exterioară sau interioară a adsorbanților minerali și de procesele de dizolvare a vaporilor sau gazelor în microporii lor, care, de regulă, nu sunt însoțite de o schimbare semnificativă. în structura sorbantului.
În timpul umflării, moleculele unui lichid cu greutate moleculară mică (sau vaporii acestuia) pătrund între elementele structurii polimerului, provocând umflarea interstructurală sau în interiorul structurilor, împingând macromoleculele în afară ( tumefacție intrastructurală). În același timp, datorită capacității macromoleculelor de a-și schimba forma, solventul nu numai că umple golul dintre legăturile individuale, dar crește și razele efective ale bobinelor polimerice și distanța dintre centrele lor de masă, fără a perturba continuitatea corpul polimeric. Acest lucru duce la o creștere a volumului și a masei corpului polimeric în comparație cu originalul. În acest caz, volumul întregului sistem (polimer + solvent) scade de obicei - fenomenul de contracție. Contracția sistemului la umflarea polimerului se explică prin orientarea moleculelor de solvent de-a lungul macromoleculelor, iar sistemul devine mai compact.
Prin urmare, procesul de umflare este sorbția (absorbția) a unei substanțe cu greutate moleculară mică de către un polimer, însoțită de o creștere a masei, volumului și modificării structurii acesteia..
Este obișnuit să se reprezinte procesul de umflare ca având loc în două etape:
Izolarea"\u003e Umflarea nelimitată este umflarea care se transformă spontan în dizolvare. Este analogă amestecării nelimitate de lichide, cum ar fi apă și alcool etilic sau apă și acid sulfuric.
Polimerul umflat, care este o soluție dintr-un lichid cu greutate moleculară mică într-un polimer, coexistă de ceva timp cu un strat de lichid pur cu greutate moleculară mică. După o anumită perioadă de timp, când lanțurile polimerice sunt deja suficient de separate, acestea încep să difuzeze încet în solvent. Apare un strat de soluție mai diluată, coexistând cu un strat de soluție mai concentrată. După ceva timp, concentrațiile ambelor straturi devin egale - straturile se îmbină, formând un sistem omogen monofazat.
Umflare limitată- procesul de interacţiune a polimerilor cu lichide cu greutate moleculară mică, limitat doar de stadiul absorbţiei acestora de către polimer; nu are loc dizolvarea spontană a polimerului; lanțurile polimerice nu sunt complet separate unele de altele.
În acest caz, se formează două faze coexistente. O fază este o soluție dintr-un lichid cu greutate moleculară mică într-un polimer, cealaltă este un lichid pur cu greutate moleculară mică (dacă polimerul nu se dizolvă deloc) sau o soluție diluată a unui polimer într-un lichid cu greutate moleculară mică. Aceste faze sunt separate printr-o interfață clar vizibilă și sunt în echilibru.
Ar trebui făcută o distincție între umflarea limitată a polimerilor liniari și de rețea. Pentru polimerii liniari, acest proces este similar cu amestecarea limitată a lichidelor: în anumite condiții (temperatura, concentrația componentelor), umflarea este limitată, dar cu o schimbare corespunzătoare a condițiilor, se poate transforma în dizolvare nelimitată. De exemplu, gelatina se umflă într-o măsură limitată în apă la temperatura camerei și, atunci când este încălzită la aproximativ 35 ° C, se dizolvă în apă pe termen nelimitat.
Dacă polimerul are o rețea spațială formată din legături chimice, atunci lanțurile nu pot fi separate la nicio temperatură (sub temperatura de descompunere a polimerului). Prin urmare, polimerii de rețea sunt fundamental insolubili, dar se pot umfla pentru a forma jeleuri sau geluri.
Gradul de umflătură și cinetica umflăturii.În practică, este foarte important să se cunoască capacitatea polimerilor de a se umfla în diverse medii lichide și de vapori. Această capacitate este măsurată prin gradul de umflare, care este exprimată prin cantitatea de lichid (sau vaporii acestuia) absorbită de polimer per unitate de masă sau volum a polimerului.
Gradul de umflare poate fi determinat în funcție de greutate sau de volum. Metoda greutății constă în cântărirea probei înainte și după umflare și în calcularea gradului de umflare formula "src="http://hi-edu.ru/e-books/xbook839/files/f202
unde exemplu "> m - o probă de polimer umflat.
Metoda volumetrică pentru determinarea gradului de umflare se bazează pe măsurarea volumului polimerului înainte și după umflare:
formula" src="http://hi-edu.ru/e-books/xbook839/files/f205.gif" border="0" align="absmiddle" alt="- volumul polimerului original; V este volumul polimerului umflat.
Gradul de umflare poate fi determinat numai pentru polimerii cu umflare limitată, deoarece cu umflare nelimitată polimerul însuși începe să se dizolve și masa probei scade. Gradul de umflare variază în timp. Selectarea dependenței "> Fig. 10.1  .
.
După cum se poate observa din figură, începând de la un anumit timp, gradul de umflătură devine constant. Mărimea gradului de umflătură, care corespunde apariției unei secțiuni orizontale pe curbă, se numește maximă sau gradul de echilibru de umflare. Pentru diferiți polimeri, gradul de echilibru de umflare este stabilit la diferite intervale de timp, ceea ce are o mare importanță practică. Deci, gradul de umflare de echilibru al unui polimer (curba 2 din Fig. 10.1) poate fi mai mare decât cel al altuia (curba 1). În consecință, atunci când sunt în acest lichid pentru o perioadă lungă de timp, a doua probă se va umfla mult mai mult.
Dacă gradul de umflare este determinat după o perioadă mai scurtă de timp, se poate observa imaginea opusă: gradul de umflare al primului eșantion va fi mai mare decât al celui de-al doilea. Pentru a evalua capacitatea unui polimer de a se umfla, ar trebui să folosiți valoarea gradul maxim de umflare(izolare"\u003e umflarea negativă a polimerului - nu o creștere, ci o scădere a masei probei în timp. Acest lucru are loc atunci când polimerii unei structuri liniare sau de rețea se umflă ca urmare a leșierii impurităților solubile din ei. Determinarea gradul de umflare este utilizat în practică la testarea produselor polimerice finite destinate lucrului în medii lichide și gazoase.
Viteza de umflare este limitată de viteza de difuzie a solventului în faza polimerului și, pentru polimerii în starea inițială foarte elastică, poate fi descrisă prin ecuația cinetică de ordinul întâi:
exemplu">k - constanta vitezei de umflare, exemplu">t .
Integrarea ecuației (10.3) dă
formula" src="http://hi-edu.ru/e-books/xbook839/files/f201.gif" border="0" align="absmiddle" alt="din timp în coordonate exemplu "> t ar trebui grupat de-a lungul unei linii drepte cu o tangentă a unghiului de înclinare egală cu k (Fig. 10.2).  ). Acest lucru arată că, din punct de vedere cinetic, procesul de umflare este o reacție de ordinul întâi.
). Acest lucru arată că, din punct de vedere cinetic, procesul de umflare este o reacție de ordinul întâi.
Cinetica umflaturii nelimitate unii polimeri este prezentat în fig. 10.3  . Până la punctul a, pentru toți polimerii, se observă o creștere treptată încetinită a gradului de umflare (datorită umflăturii mai intense la începutul procesului). La punctul a, viteza de dizolvare devine egală cu rata de umflătură, iar de ceva timp gradul de umflătură nu se modifică. La punctul b, viteza de dizolvare începe să depășească viteza de umflare, iar greutatea probelor scade. Între punctele a și b, probele au formula pentru gradul maxim de umflare .
. Până la punctul a, pentru toți polimerii, se observă o creștere treptată încetinită a gradului de umflare (datorită umflăturii mai intense la începutul procesului). La punctul a, viteza de dizolvare devine egală cu rata de umflătură, iar de ceva timp gradul de umflătură nu se modifică. La punctul b, viteza de dizolvare începe să depășească viteza de umflare, iar greutatea probelor scade. Între punctele a și b, probele au formula pentru gradul maxim de umflare .
Dintr-o comparație a curbelor de umflare, putem concluziona că, cu cât greutatea moleculară este mai mică, cu atât este mai mică ramificarea macromoleculelor și interacțiunea intermoleculară și cu cât afinitatea termodinamică dintre polimer și solvent este mai mică, cu atât formula „src="http este mai mică. ://hi-edu.ru/e-books/ xbook839/files/f211.gif" border="0" align="absmiddle" alt=".
Cu o greutate moleculară foarte mare sau o interacțiune intermoleculară puternică, unii polimeri (de exemplu, proteinele) se dizolvă extrem de lent (curba 3 din Fig. 10.3) și pot menține gradul maxim de umflare pentru o perioadă lungă de timp, adică. caracterizată prin umflare limitată. Dizolvarea unor astfel de polimeri umflați necesită agitare, creșterea temperaturii; în acest caz, interacțiunea intermoleculară este slăbită și mobilitatea macromoleculelor crește, ceea ce accelerează dizolvarea acestora.
Cinetica umflaturii limitate. După cum sa menționat deja, umflarea limitată este procesul de interacțiune a polimerilor cu lichide cu greutate moleculară mică, care nu este însoțită de dizolvare. Acest lucru se observă la o afinitate termodinamică scăzută a polimerului și solventului și este, de asemenea, caracteristic polimerilor ale căror macromolecule sunt conectate prin legături încrucișate puternice într-o rețea tridimensională.
Legăturile încrucișate rare între macromolecule în prima etapă a umflării polimerului nu împiedică difuzia moleculelor de solvent în acesta. Prin urmare, în prima perioadă, umflarea apare într-un ritm maxim. Cu toate acestea, solvatarea solventului prin legături macromoleculare situate între nodurile rețelei reduce mobilitatea acestora, duce la creșterea distanțelor dintre ele, la întinderea și îndreptarea macromoleculelor, la scăderea entropiei sistemului, la apariția unor puternice mecanice. tensiuni si ruperea unor sectiuni suprasolicitate; rata de umflare scade. (vezi fig. 10.1 și fig. 10.4  ).
).
Cu o creștere a numărului de legături încrucișate, de ex. densitatea rețelei spațiale, gradul și rata de umflătură sunt reduse. Cinetica umflării limitate a polimerilor de rețea este prezentată în fig. 10.5  .
.
Forma particulară a curbei 3 este explicată prin extracția componentelor solubile cu greutate moleculară mică ale amestecului din polimerul rețelei de umflare și dizolvarea acestora. În secțiunea a - b, vitezele de umflare și dizolvare sunt aceleași, în secțiunea b - c, prevalează dizolvarea, în secțiunea c - d, dizolvarea se termină..gif" border="0" align="absmiddle" alt="( !LANG:- umflarea maximă a polimerului rețelei, ">
Natura chimică a polimerului și a solventului;
greutatea moleculară a polimerului;
Flexibilitatea lanțului polimeric;
Densitatea de ambalare a macromoleculelor;
Starea de fază a polimerului;
Eterogenitatea compoziției chimice a lanțului;
Prezența și frecvența rețelei spațiale;
temperatura.
Să le luăm în considerare.
Natura polimerului și solventului. Solubilitatea reciprocă a substanțelor depinde de structura lor chimică. Din timpuri imemoriale, a existat o poziție: „ca se dizolvă în asemănător”, adică. substanțele care sunt similare ca structură chimică se dizolvă reciproc, iar substanțele care diferă puternic în structură chimică nu se dizolvă. Ulterior, apropierea energiei interacțiunii intermoleculare a fost luată ca semn de „similar”.
Să luăm în considerare soluțiile în care apar doar interacțiunile van der Waals, i.e. interacțiuni de dispersie, inducție și orientare.
Dacă lichidele sunt nepolare, de ex. au un moment dipol zero și doar forțele de dispersie acționează între ele, de obicei se amestecă bine între ele pe o gamă largă de temperaturi. Din acest punct de vedere, se poate aștepta o bună dizolvare a hidrocarburilor polimerice amorfe nepolare (poliizobutilenă, poliizopren, polibutadienă etc.) în hidrocarburi saturate cu greutate moleculară mică și în amestecul acestora (benzină), ceea ce se observă de fapt. Astfel de polimeri nu se dizolvă sau se umflă în lichide polare (acetonă etc.) și mai ales în lichide capabile să formeze legături de hidrogen (apă, alcooli inferiori).
Dimpotrivă, polimerii care conțin grupări polare (nitrat de celuloză, poliacrilonitril etc.) nu se dizolvă în lichide nepolare și au tendința de a interacționa cu lichidele apropiate acestora în polaritate.
Polaritatea unei substanțe este de obicei caracterizată de mărimea momentului dipol permanent, care, totuși, nu este o măsură eficientă a polarității. De exemplu, toți alcoolii alifatici sau toate cetonele au aproape același moment dipol și putere de solvent complet diferită față de alte lichide și polimeri. Pentru aceeași valoare a momentului dipol, cu cât radicalul de hidrocarbură este mai lung într-o moleculă de alcool sau cetonă, cu atât polimerii nepolari mai buni se dizolvă sau se umflă sau cu atât polimerii polari mai rău se dizolvă sau se umflă.
Toate acestea mărturisesc natura extrem de complexă a interacțiunii polimerilor cu solvenții, care depinde atât de forma și dimensiunea macromoleculelor, cât și de capacitatea acestora de a suferi transformări conformaționale, greutate moleculară, densitate de împachetare și alți factori.
Greutatea moleculară a polimerilor. Odată cu creșterea greutății moleculare a polimerului în orice serie de omologie a polimerului, capacitatea de dizolvare scade întotdeauna. Membrii cu greutate moleculară mică ai seriei se pot dizolva la infinit într-un lichid dat, în timp ce membrii cu greutate moleculară mare se pot umfla doar într-o măsură limitată. Acest lucru se datorează energiilor mari de interacțiune care se însumează de-a lungul întregii lungimi a lanțului și mobilității limitate a legăturilor de lanț interconectate prin legături chimice. Diferitele proprietăți de dizolvare ale omologilor polimerilor sunt folosite pentru a le separa în fracții.
Flexibilitatea lanțului polimeric. Mecanismul dizolvării polimerului constă în separarea lanțurilor unele de altele și difuzarea lor în faza solventului. Acest lucru este facilitat de flexibilitatea lanțului. Un lanț flexibil se poate deplasa în părți, legăturile sale sunt capabile să facă schimb de locuri cu molecule de solvent, difuzia sa se realizează prin mișcarea secvențială a grupurilor de legături, care nu necesită cheltuieli mari de energie pentru a depăși interacțiunile intermoleculare. Prin urmare, polimerii amorfi cu lanțuri flexibile, de regulă, se umflă la nesfârșit; dizolva.
Lanțurile de polimeri nepolari, care sunt capabili să interacționeze cu lichide nepolare, au o mare flexibilitate. Prin urmare, polimerii amorfi nepolari cu lanțuri flexibile în orice lichid nepolar sunt practic solubili nelimitat.
Lanțurile rigide se pot mișca doar în întregime, ceea ce, în prezența unei greutăți moleculare mari, face difuzarea lor foarte dificilă. Legăturile lor nu pot face schimb independent de locuri cu molecule de solvent. Prin urmare, polimerii liniari amorfi, ale căror lanțuri sunt rigide datorită prezenței grupărilor polare, se umflă bine în lichide extrem de polare, dar, de regulă, nu se dizolvă în ele la temperaturi obișnuite. Pentru a dizolva polimeri foarte rigizi, este necesară o interacțiune puternică între polimer și solvent. De exemplu, celuloza se dizolvă în baze de amoniu cuaternar, adică. în lichide cu care poate forma complexe.
Densitatea de ambalare a macromoleculelor. Energia interacțiunii intermoleculare este invers proporțională cu distanța până la a șasea putere. Prin urmare, cele mai mici modificări ale distanțelor, adică. în densitatea de ambalare a macromoleculelor poate schimba drastic energia de interacțiune și, în mod natural, ambalarea mai slabă ar trebui să îmbunătățească solubilitatea, iar ambalarea mai densă ar trebui să o agraveze.
Atât lanțurile foarte flexibile, cât și cele foarte rigide se pot împacheta strâns, dar acest lucru afectează solubilitatea în moduri diferite. Polimerii nepolari amorfi cu lanț flexibil, în ciuda ambalării dense, sunt foarte solubili în lichide nepolare datorită posibilității de mișcare segmentară. Polimerii cu catenă rigidă strâns ambalați nu au această capacitate și, prin urmare, sunt foarte greu de dizolvat. Acest lucru este tipic pentru astfel de poliamide aromatice ca poli-n-benzamidă, poli-n-tereftalamidă și alte poliheteroarilene strâns împachetate.
Starea de fază a polimerului. Deoarece densitatea de ambalare a macromoleculelor afectează solubilitatea, este clar că prezența unei rețele cristaline joacă un rol uriaș, a cărei distrugere necesită multă energie. Prin urmare, polimerii cristalini, chiar nepolari, la temperatura camerei, nu se dizolvă în lichide apropiate de ei în polaritate (polietilenă, polipropilenă etc.). Pentru a dizolva polimerii cristalini, aceștia trebuie încălziți la temperaturi apropiate de punctele lor de topire. Politetrafluoretilena nu se dizolvă în niciun solvenți într-un interval larg de temperatură.
Eterogenitatea compoziției chimice. Mulți polimeri, în funcție de condițiile preparării lor, pot avea o compoziție chimică diferită. De exemplu, acetații de celuloză pot avea grade diferite de acetilare, nitrații de celuloză pot avea grade diferite de nitrare, probele de alcool polivinilic industrial conțin adesea un număr diferit de grupări acetil. Astfel de polimeri au solubilitate diferită. De exemplu, triacetatul de celuloză este solubil în clorură de metilen, acid acetic glacial și acid formic, dar are o miscibilitate limitată cu cetone și esteri. Acetatul de celuloză, care conține aproximativ 54-57% grupări acetil, este miscibil liber cu acetona și alte cetone. Azotatul de celuloză, care conține 10-12% azot, se amestecă fără restricții cu acetona, iar trinitratul de celuloză se umflă doar puțin în el. În acest caz, ambii nitrați nu interacționează cu apa și hidrocarburile.
legături chimice încrucișate. Chiar și un număr mic de legături chimice încrucișate între lanțuri le împiedică să se separe unul de celălalt și să intre în soluție. Pentru a obține un polimer insolubil, este suficient să se creeze cel puțin o legătură între lanțurile adiacente; de exemplu, în vulcanizarea cauciucurilor cu sulf, 2 moli de polimer necesită 1 mol de sulf. Aceasta înseamnă că, cu o greutate moleculară medie a cauciucului egală cu 100.000, sunt necesare 32 g de sulf la 200.000 g de cauciuc sau aproximativ 0,16 g de sulf la 1 kg de cauciuc. Dacă cauciucul este capabil de reacții de reticulare atunci când interacționează cu oxigenul, atunci prezența a 0,08 g de oxigen la 1 kg de cauciuc este suficientă pentru ca cauciucul să nu se dizolve. Astfel, cantitățile neglijabile de aditivi de reticulare privează complet polimerii de capacitatea de a se dizolva în orice solvenți. Polimerii reticulati nu devin solubili atunci cand sunt incalziti la orice temperatura.
Dacă numărul de legături încrucișate din polimer este relativ mic, de ex. segmentele de lanț dintre ele sunt suficient de mari, apoi moleculele de substanțe cu greutate moleculară mică pot pătrunde în faza polimerică. Această pătrundere este însoțită de separarea segmentelor de lanțuri adiacente; prin urmare, polimerul reticulat se poate umfla într-o măsură limitată. În acest caz, gradul de umflare depinde de toți factorii discutați mai sus și de frecvența rețelei spațiale. Deci, polimerii de rețea nepolari se umflă mai bine în lichidele nepolare, cele polare - în cele polare. Polimerii având grupări capabile să formeze legături de hidrogen se umflă în lichide cu care pot forma aceste legături. Ochiurile flexibile sau slab împachetate, celelalte lucruri fiind egale, se umflă mai bine decât ochiurile rigide sau dens.
O creștere a numărului de legături încrucișate duce la o scădere a capacității polimerului de a absorbi un lichid cu greutate moleculară mică; în prezența unei rețele spațiale dense, polimerul își pierde complet capacitatea de a se umfla. Acest lucru poate fi ilustrat cu o serie de exemple.
Astfel, odată cu creșterea conținutului de sulf din polimer, capacitatea vulcanizatelor de cauciuc de a se umfla în mod continuu scade. Ebonita (un vulcanizat care conține aproximativ 32% sulf) nu se umflă deloc. Polimerii rezolu fenol-formaldehidă cu o structură liniară sunt ușor solubili în acetonă și alcool. Resitol, adică produsul unei structuri de rețea, se umflă doar într-o măsură limitată în acești solvenți, iar resit este complet lipsit de capacitatea de a se umfla.
Temperatura. Solubilitatea polimerilor se poate îmbunătăți și deteriora odată cu creșterea temperaturii, care depinde de efectul temperaturii asupra afinității termodinamice a sistemului polimer-solvent. Acest lucru rezultă în mod clar din luarea în considerare a echilibrului de fază al sistemelor polimerice.
Principala lege de echilibru a unui sistem multifazic multicomponent este regula fazei Gibbs, care stabilește relația dintre numărul de faze Ф, numărul de componente din sistemul K și numărul gradelor sale de libertate.
formula" src="http://hi-edu.ru/e-books/xbook839/files/f216.gif" border="0" align="absmiddle" alt="
Conform ecuației (10.6), un sistem condensat monofazat cu două componente are două grade de libertate (starea sistemului este determinată de temperatura și concentrația uneia dintre componente).
În prezența a două faze (r = 2), sistemul bicomponent condensat are un grad de libertate. Aceasta înseamnă că o modificare a temperaturii determină o modificare a concentrației ambelor faze. La o anumită temperatură, aceste faze se pot îmbina pentru a forma o soluție omogenă monofazată. În schimb, o soluție omogenă monofazată la o anumită temperatură se poate delamina sau se poate separa în două faze. Temperatura la care are loc separarea se numește temperatura de separare a fazelor, sau separarea fazelor(formula" src="http://hi-edu.ru/e-books/xbook839/files/f217.gif" border="0" align="absmiddle" alt=", a cărui dependență de compoziția soluției se exprimă curba de amestecare, sau curba de frontieră separând regiunea soluţiilor monofazate de cele bifazate.
Primul semn de separare a unui sistem transparent omogen în două faze, sau formarea unei noi faze, este o ușoară tulburare a sistemului (opalescență). Acesta este rezultatul împrăștierii luminii de către cele mai mici particule din faza nou formată. Aceasta se bazează pe o metodă de obținere a diagramelor de stare, care este numită metoda punctului de nor. Constă în încălzirea și răcirea succesivă foarte lentă a soluțiilor de o concentrație dată și înregistrarea formulei „alt="din compoziție, care este exprimată în moli, fracțiuni de masă sau volum ale componentelor.
Diagramele de fază cunoscute pentru sistemele HMS-solvent sunt prezentate în fig. 10.6  . Deoarece masa molară a HMS este mult mai mare decât masa molară a solventului, compoziția din aceste diagrame este de obicei exprimată în masă sau volum, dar nu în fracții molare. Sisteme cunoscute cu temperatura de dizolvare critică superioară (UCST), peste care polimerul este complet dizolvat într-un lichid cu greutate moleculară mică la orice concentrație. Zona de deasupra curbei corespunde unui sistem omogen monofazat, zona de sub curbă corespunde unui sistem eterogen bifazic. De exemplu, la punctul C (Fig. 10.6, a) sistemul este stratificat în două faze de echilibru ale compozițiilor formula „src="http://hi-edu.ru/e-books/xbook839/files/f219.gif" border="0 " align="absmiddle" alt="(soluție saturată de solvent în polimer).
. Deoarece masa molară a HMS este mult mai mare decât masa molară a solventului, compoziția din aceste diagrame este de obicei exprimată în masă sau volum, dar nu în fracții molare. Sisteme cunoscute cu temperatura de dizolvare critică superioară (UCST), peste care polimerul este complet dizolvat într-un lichid cu greutate moleculară mică la orice concentrație. Zona de deasupra curbei corespunde unui sistem omogen monofazat, zona de sub curbă corespunde unui sistem eterogen bifazic. De exemplu, la punctul C (Fig. 10.6, a) sistemul este stratificat în două faze de echilibru ale compozițiilor formula „src="http://hi-edu.ru/e-books/xbook839/files/f219.gif" border="0 " align="absmiddle" alt="(soluție saturată de solvent în polimer).
Sistemele cu VCTR sunt: acetat de celuloză - cloroform, poliizobutilenă - benzen, polistiren - ciclohexan etc. În fig. 10.6, b prezintă o diagramă a unui sistem cu o temperatură critică de dizolvare (LCST) mai mică, sub care polimerul și solventul sunt infinit solubile unul în celălalt. De exemplu, sistemele oxid de polietilenă - apă, metilceluloză - apă, nitrat de celuloză - etanol au LCST. Pentru unele sisteme (oxid de polipropilenă - apă), se realizează curbe de solubilitate închise cu LCST și VCST - în fig. 10.6, în .
Este cunoscut un alt tip de diagrame de fază, pentru care LCST este deasupra VKTR și deasupra punctului de fierbere al solventului (Fig. 10.6, d). Astfel de diagrame sunt caracteristice sistemelor al căror polimer și solvent sunt apropiate ca structură chimică. În acest caz, LCTE crește odată cu creșterea dimensiunii moleculelor de solvent. Stratificarea sistemului în acest caz se explică de obicei printr-o diferență mare a coeficienților de dilatare termică a componentelor. Stați diagrame de tipul prezentat în fig. 10,6, g obținut pentru sistemele polietilenă - alcani, acetat de polivinil - acetat de etil, alcool polivinilic - apă etc.
Termodinamica soluțiilor explică existența diagramelor prezentate în fig. 10.6 după cum urmează. Deoarece soluțiile de polimeri se formează spontan, formarea lor, ca orice proces spontan, este însoțită de o scădere a energiei Gibbs, adică.
formula" src="http://hi-edu.ru/e-books/xbook839/files/f221.gif" border="0" align="absmiddle" alt=".gif" border="0" align="absmiddle" alt="este suma energiilor Gibbs ale componentelor înainte de dizolvare.
Energia Gibbs este legată de modificarea entalpiei și entropiei procesului prin ecuație
formula" src="http://hi-edu.ru/e-books/xbook839/files/f225.gif" border="0" align="absmiddle" alt=".gif" border="0" align="absmiddle" alt="Rezultă că dizolvarea spontană a polimerului fără delaminarea sistemului se realizează în mai multe moduri.
1. def"> și def">2. def">și def">supus formulei" src="http://hi-edu.ru/e-books/xbook839/files/f234.gif" border="0" align="absmiddle" alt="( !LANG:.gif" border="0" align="absmiddle" alt="< 0, выше НКТР опред-е">3. def "> și def"> supuse def"> 4. def"> și selecție"> Fig. 10.6 corespund cazurilor de formula de dependență complexă" src="http://hi-edu .ru/e-books /xbook839/files/f238.gif" border="0" align="absmiddle" alt=".gif" border="0" align="absmiddle" alt="mai mare decât zero, în timp ce altele sunt mai mici.
În formarea soluţiilor ideale(supunând legii lui Raoult) formula" src="http://hi-edu.ru/e-books/xbook839/files/f242.gif" border="0" align="absmiddle" alt="= 0 (volumul soluției este egal cu suma volumelor componentelor). În acest caz, dizolvarea se datorează doar unei creșteri a entropiei. Entropia, pe de altă parte, crește datorită creșterii numărului de microstări echivalente în energie în procesul de amestecare..gif" border="0" align="absmiddle" alt=".gif" border="0" align="absmiddle" alt="< 0.
Soluțiile polimerice prezintă întotdeauna abateri semnificative de la comportamentul ideal, chiar dacă formula =". adică..gif" border="0" align="absmiddle" alt=".gif" border="0" align="absmiddle" alt="soluția atermică a introdus un membru suplimentar care conține o formulă a parametrilor fără dimensiune" src="http://hi-edu.ru/e-books/xbook839/files/f235.gif" border="0" align="absmiddle" alt=" solutii reale:
formula" src="http://hi-edu.ru/e-books/xbook839/files/f249.gif" border="0" align="absmiddle" alt=".gif" border="0" align="absmiddle" alt=".gif" border="0" align="absmiddle" alt="> 1/2..gif" border="0" align="absmiddle" alt="este o măsură a afinității termodinamice a unui solvent pentru un polimer sau o măsură a calității unui solvent (pentru solvenți foarte buni poate fi mai mică decât zero).
Capacitatea de a umfla și dizolva polimerii în diverși solvenți depinde de structura moleculelor acestora. Regula generală „ca se dizolvă asemănător” este susținută de faptul că, în general, polimerii nepolari se dizolvă ușor în solvenți nepolari și nu se dizolvă în cei polari; dimpotrivă, polimerii polari nu se dizolvă în solvenți nepolari, dar se dizolvă în cei polari.
Umflarea și solubilitatea unui polimer într-un anumit solvent depind de interacțiunile grupărilor funcționale sau ale atomilor, care au ca rezultat legături donor-acceptator și alte legături care conduc la formarea de complexe stabile de macromolecule polimerice cu molecule de solvent.
Procesul de dizolvare (umflare) are loc numai dacă componentele pot fi amestecate sau dizolvate reciproc, adică. depinde dacă între ele există o afinitate termodinamică. În funcție de gradul de afinitate termodinamică a solvenților pentru polimeri, aceștia se împart în termodinamic compatibili cu polimeri și incompatibili.
Evaluarea cantitativă a afinității termodinamice a componentelor unul față de celălalt se realizează în funcție de gradul de reducere ca urmare a interacțiunii potențialelor lor chimice, formula "src="http://hi-edu.ru/e -books/xbook839/files/f252.gif" align="absmiddle" alt=".gif" border="0" align="absmiddle" alt="
Potențialul chimic al unei componente în soluție formula" src="http://hi-edu.ru/e-books/xbook839/files/f256.gif" border="0" align="absmiddle" alt="(! LANG:.gif" border="0" align="absmiddle" alt="și def-e">definition selection">osmotic .
Presiunea osmotică este un indicator al afinității termodinamice dintre componente, pe unitatea de volum de solvent: cu cât presiunea osmotică este mai mare, cu atât valoarea absolută a formulei !lang:
unde c este concentrația substanței dizolvate; R este constanta universală a gazului; T este temperatura.
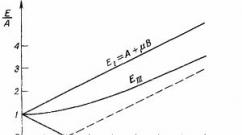
În soluțiile de polimeri, fiecare segment al macromoleculei se mișcă independent de celelalte și se comportă ca o unitate independentă cinetic. Prin urmare, presiunea osmotică a soluțiilor polimerice nu respectă legea van't Hoff și depinde de flexibilitatea macromoleculelor: cu cât flexibilitatea este mai mare, cu atât segmentele sunt mai scurte și cu cât sunt conținute mai multe în macromoleculă, cu atât presiunea osmotică este mai mare. și cu atât abaterea de la legea van't Hoff este mai puternică.
În plus, presiunea osmotică a soluțiilor de polimer crește nu proporțional cu concentrația acestora, ca în cazul substanțelor cu greutate moleculară mică, ci mult mai rapid (Fig. 10.7).
formula" src="http://hi-edu.ru/e-books/xbook839/files/f323.gif" border="0" align="absmiddle" alt="din concentrația C a soluției
Prin urmare, pentru a descrie dependența presiunii osmotice de concentrația polimerilor, ecuația van der Waals este utilizată sub forma unei expansiuni virale, mai degrabă decât ecuația van't Hoff, adică, prin grade de concentrare cu:
formula" src="http://hi-edu.ru/e-books/xbook839/files/f263.gif" border="0" align="absmiddle" alt="
unde c este concentrația polimerului în soluție; formula" src="http://hi-edu.ru/e-books/xbook839/files/f265.gif" border="0" align="absmiddle" alt=".
Primul coeficient virial este formula" src="http://hi-edu.ru/e-books/xbook839/files/f266.gif" border="0" align="absmiddle" alt="= 1/M. Al treilea și următorii termeni ai seriei de puteri au un efect redus asupra rezultatelor, astfel încât ecuația (10.13) poate fi scrisă ca
formula" src="http://hi-edu.ru/e-books/xbook839/files/f265.gif" border="0" align="absmiddle" alt="este exprimat printr-o serie de linii drepte în formă de evantai (Fig. 10.8), a căror panta este determinată de valoarea celei de-a doua formule a coeficientului virial "src="http://hi-edu.ru/e-books/ xbook839/files/ris10.8.gif" border="0 " align="absmiddle" alt="
Orez. 10.8. Dependența presiunii osmotice de concentrația soluțiilor de polimeri care diferă în valoarea celui de-al doilea coeficient virial alt=".gif" border="0" align="absmiddle" alt=">0 ("bună" afinitate termodinamică)
Cu cât valoarea pozitivă a formulei este mai mare" src="http://hi-edu.ru/e-books/xbook839/files/f268.gif" border="0" align="absmiddle" alt="(!LANG :.gif" border="0" align="absmiddle" alt="= 0 și ecuația (10.12) intră în ecuația van't Hoff (10.11).
Din Tabel. 10.1 arată, de exemplu, că la temperaturi date, dicloroetanul este un solvent bun pentru polistiren, iar ciclohexanul este unul rău. În același timp, ciclohexanul este un solvent bun pentru poliizobutilenă.
Tabelul 10.1.
Valorile celui de-al doilea coeficient virial pentru unele sisteme polimer-solvent
Document fără nume
Polimer |
M 10 -3 |
Solvent |
Temperatura, °C |
A 2 10 -4,
|
| Polistiren | ||||
Dicloroetan |
||||
| orez. 10.9 fig. Soluțiile 10.9, b sau gelurile polimerice au o importanță deosebită în industria alimentară și fotografică, în industria fibrelor artificiale și sintetice și a produselor din cauciuc și într-o serie de alte ramuri ale tehnologiei. Un jeleu sau gel este un sistem binar format dintr-o rețea spațială formată din macromolecule sau agregatele acestora, în care sunt distribuite moleculele unui lichid cu greutate moleculară mică. Astfel de rețele se pot forma și în soluții la concentrații suficient de mari de polimeri. Principala diferență dintre jeleu și soluție este că în soluții astfel de rețele au un caracter de fluctuație, adică. sunt distruse și formate continuu sub influența mișcării termice; în jeleu, grila are caracter de nefluctuație, adică. in aceste conditii, este stabil si nu se prabuseste sub actiunea miscarii termice. Dacă, odată cu schimbarea temperaturii, legăturile încrucișate pot fi distruse prin mișcarea termică, de ex. grila capata un caracter de fluctuatie, jeleul trece in stare de solutie. Acest proces se numește jeleu care se topește. Există două tipuri de știfturi. Jeleul de tip I este un sistem lichid de polimer cu greutate moleculară mică (LMW) în care rețeaua spațială este formată prin legături chimice între molecule. Aceste legături nu sunt distruse atunci când sunt încălzite, iar astfel de jeleuri nu se topesc la nicio temperatură. Încălzirea acestor jeleuri peste o anumită temperatură duce, din cauza distrugerii termice, la distrugerea ireversibilă a întregului sistem. Prin urmare, astfel de jeleuri sunt numite termo-ireversibile. Se formează jeleuri de tip I: Cu umflarea spontană a polimerilor reticulați spațial; Cu polimerizare tridimensională sau policondensare în soluție; În procesul de reticulare chimică reacții care au loc în prezența unui solvent. Jeleurile formate prin legături chimice nu sunt capabile să curgă, deoarece macromoleculele, fiind reticulate, nu se pot deplasa unele față de altele. Starea în care sistemul își pierde fluiditatea în procesul de polimerizare sau policondensare în soluție și trece dintr-o masă vâscoasă într-un jeleu se numește punct de gel, sau punct de gel . Un exemplu de jeleu de tip I sunt vulcanizate de cauciuc umflate, copolimeri de rețea umflați stiren și divinilbenzen si altele.Umflarea lor apare spontan, rezultand formarea de jeleuri monofazate termodinamic stabile de echilibru. Gradul de echilibru de umflare depinde de gradul de reticulare a polimerului, de temperatură și de natura solventului. Jeleurile de tip II sunt sisteme în care rețeaua spațială este formată din legături intermoleculare de natură variată. În anumite condiții, aceste legături sunt stabile, dar atunci când condițiile se schimbă (temperatura, natura NMF etc.), ele se pot dezintegra. În acest caz, se formează o soluție adevărată omogenă. Când sistemul revine la condițiile sale originale, se formează din nou legături puternice între lanțuri - sistemul devine gelatinos. Astfel de studenți sunt chemați termoreversibilă. Jeleurile de tip II se formează prin interacțiunea polimerilor liniari sau ramificati având grupări amfifile (diferite ca polaritate) cu solvenți nu prea buni în sens termodinamic, care interacționează doar cu un tip de grup și nu interacționează cu altul. Într-un mediu cu un solvent foarte bun pentru un anumit polimer, jeleul, de regulă, nu se formează. Prin urmare, formarea gelului are loc atunci când condițiile se schimbă, ceea ce duce la o deteriorare a puterii de dizolvare a mediului. Acest lucru se poate întâmpla atunci când temperatura se schimbă sau când un solvent rău este adăugat la o soluție de polimer într-un solvent bun. Acesta este un sistem de neechilibru, a cărui stare de echilibru corespunde formării a două faze. Prin urmare, jeleul de tip II este bifazic. Clasic exemplu jeleul bifazic sunt jeleuri de gelatină, formate prin răcirea soluțiilor sale. O rețea stabilă de fluctuații în ele, aparent, este creată de legăturile dintre situsurile de hidrocarburi hidrofobe și de legături puternice de hidrogen între grupările -NH-C ale lanțurilor adiacente, pe care apa nu le distruge la temperaturi obișnuite. Jeleurile cu două faze se formează atunci când soluțiile sunt răcite poliacrilonitril în dimetilformamidă etc. Dacă polimerul este capabil să cristalizeze, atunci jeleul rezultat are o structură cristalină. Acest lucru se observă, de exemplu, la răcirea soluțiilor alcool polivinilic în etilen glicol sau glicerină, într-o măsură mai mică - la răcirea soluțiilor apoase Alcool polivinil. Cristalizarea este întotdeauna cu atât mai perfectă, cu atât natura chimică a NMF și a legăturii polimerului este mai apropiată. Jeleurile termoreversibile sunt formate prin răcirea soluțiilor de polimeri de tip pieptene cristalizați, un număr de poliacrilați în alcooli alifatici și hidrocarburi. Legăturile stabile în acest caz apar datorită interacțiunii ramurilor de metilen la o lungime suficient de mare, ceea ce creează o ordine de orientare și duce la cristalizare. Următorii factori influențează procesul de formare a gelului: concentrația DIU în soluție; Forma și dimensiunea moleculelor DIU; Temperatura; Prezența electroliților indiferenți pH-ul mediului. Efectul concentrației DIU Gelificarea soluțiilor HMS este întotdeauna facilitată de o creștere a concentrației soluției, deoarece aceasta crește frecvența coliziunilor între macromolecule sau regiunile acestora și crește numărul de legături formate pe unitate de volum. Influența formei și dimensiunii macromoleculelor Macromoleculele nu sunt doar mari, dar, și acest lucru este foarte important, au flexibilitatea lanțurilor polimerice, ceea ce oferă capacitatea de a prelua un număr mare de conformații: de la o stare absolut întinsă la o bobină strânsă. Desigur, numărul de legături pe care o anumită macromoleculă le formează cu altele depinde de forma moleculei: cu cât este mai îndreptată, cu atât este mai ușor să accesezi acele părți ale acesteia care pot interacționa. Prin urmare, pentru formarea gelului, sunt necesare condiții în care macromolecula să nu se coaguleze într-o bilă. Macromoleculele având o formă alungită formează jeleu chiar și în soluții foarte diluate. Deci, agar-agar formează un jeleu la un conținut de 0,1%, iar gelatina - 0,5% substanță uscată. La meduzele de mare, care sunt jeleuri „vii”, cantitatea de apă ajunge la 99%. Efectul temperaturii O creștere a temperaturii, cu excepția cazului în care apar modificări chimice ireversibile în sistem, previne de obicei gelificarea datorită creșterii intensității mișcării termice a segmentelor și ca urmare a scăderii numărului și a duratei de existență a legăturilor dintre macromolecule. Când temperatura se schimbă, poate apărea gelificarea spontană a soluției de DIU adevărat. Deci, o soluție apoasă 30% de geluri de gelatină la o temperatură de 30 ° C, o soluție mai diluată de 10% necesită o temperatură mai scăzută - 22 ° C pentru gelatinizare. Trebuie remarcat faptul că trecerea soluției DIU în jeleu cu o schimbare a temperaturii are loc continuu, adică. în acest caz nu există o temperatură de tranziție constantă, așa cum este cazul, de exemplu, în timpul cristalizării sau topirii. Influența timpului Întrucât procesul de gelificare nu este altceva decât procesul de apariție și de întărire treptată a rețelei spațiale, timpul joacă cu siguranță un rol pozitiv. Cu toate acestea, nu trebuie să credeți că procesul de gelificare va avea loc în orice soluție și în orice condiții, dacă se desfășoară o perioadă lungă de timp, trebuie îndeplinite și alte condiții. Cu toate acestea, dacă jeleul este obținut ca urmare a umflării limitate a DIU uscat, atunci, în funcție de natura polimerului și a solventului, este necesar un timp foarte specific. Astfel, este nevoie de 35-40 de minute pentru a umfla gelatina în apă rece. Influența electroliților indiferenți Efectul electroliților asupra gelificării soluțiilor de proteine este opus efectului acestor electroliți asupra umflăturii. Ionii care cresc umflarea încetinesc gelificarea sau o fac imposibilă. Dimpotrivă, ionii care reduc volumul jeleului umflat contribuie la gelificare. La fel ca umflarea, gelificarea este afectată în principal de anioni. Efectul pH-ului Efectul pH-ului este vizibil mai ales dacă HMC este amfoter, cum ar fi o proteină. Gelificarea se desfășoară cel mai bine la o valoare a pH-ului corespunzătoare punctului izoelectric, deoarece în acest caz același număr de grupări ionizate încărcate opus sunt situate de-a lungul întregii lungimi a lanțului molecular, ceea ce contribuie la formarea de legături între macromoleculele individuale. în pH (în ambele direcții de la punctul izoelectric), macromoleculele capătă sarcini asemănătoare, ceea ce împiedică formarea legăturilor. Când se adaugă cantități mari de acid sau bază, gradul de ionizare scade și tendința de gelificare crește din nou. Jeleurile și gelurile au proprietăți atât solide, cât și lichide. Ca solide, au proprietăți mecanice precum elasticitatea, rezistența, elasticitatea și capacitatea de a menține o anumită formă. Spre deosebire de geluri, majoritatea jeleurilor nu sunt tixotrope. Acest lucru se datorează faptului că în jeleuri rețeaua spațială este formată din legături chimice sau de hidrogen puternice. Dacă aceste legături sunt rupte ca urmare a acțiunii mecanice, ele nu vor fi restaurate, deoarece compoziția se va modifica la punctele de rupere din cauza interacțiunii cu solventul. Într-o oarecare măsură, tixotropia poate fi observată numai în acele jeleuri, care se caracterizează printr-o rezistență scăzută a legăturilor între macromolecule. Jeleul conține o cantitate mare de apă, astfel încât acestea prezintă unele proprietăți ale lichidelor, în special substanțele cu greutate moleculară mică, solurile foarte dispersate, precum și soluțiile DIU cu molecule mici sunt capabile să difuzeze în jeleu. Pentru jeleuri, sinereza este caracteristică - o compresie treptată a rețelei spațiale cu eliberarea de lichid. Lichidul care umple plasa de jeleu este adesea denumit intermicelar, poate fi împărțit în liber, care este inclus mecanic în cadrul jeleului și nu este inclus în învelișul de solvat și legat. Cantitatea de apă legată din jeleu depinde de gradul de hidrofilitate al macromoleculei: cu cât este mai mare numărul de grupări hidrofile, cu atât mai multă apă legată în jeleu. Apa legată are proprietăți speciale: densitate mare, punct de îngheț scăzut etc. Apa legată de jeleu joacă un rol important: prezența ei în sol, plante, în toate organismele vii asigură rezistență la îngheț, menține „rezervele de apă”, determină structurile morfologice ale celulelor și țesuturilor. În corpul uman, proporția de apă legată la sugari este de aproximativ 70%, iar la vârstnici - până la 40%, ceea ce provoacă riduri, piele lăsată etc. Sinereza, prin urmare, în corpul uman este destul de lentă și viteza sa este individuală. Trebuie remarcat faptul că în timpul sinerezei, apa liberă este mai întâi eliberată și apoi, parțial, legată. Conductivitatea electrică a jeleului este apropiată de conductivitatea soluțiilor din care sunt obținute. Jeleurile, cum ar fi soluțiile și soluțiile DIU, împrăștie lumina incidentă. Jeleurile au o proprietate uimitoare ca „memoria”. Dacă uscați două jeleuri la temperatură scăzută la același conținut de umiditate, dintre care unul a fost obținut dintr-o soluție diluată și celălalt dintr-o soluție concentrată de gelatină și apoi le lăsați să se umfle din nou în apă, atunci primul jeleu se va umfla mult mai mult decât al doilea. Motivul pentru acest fenomen este că atunci când este uscat într-o anumită măsură, structura internă care a apărut în timpul formării lor se păstrează în jeleuri. Semnificația practică a stării gelatinoase este foarte mare. Pe langa cazul produselor de turnare din solutii polimerice, formarea jeleului joaca un rol extrem de important in prelucrarea produselor alimentare, in special pentru a da forma finala produselor finite. În biologie, starea gelatinoasă stă la baza proceselor de transformare a substanțelor în organisme. Multe părți constitutive ale organismelor se află într-o stare de echilibru mobil cu mediul acvatic, iar comportamentul lor este în mare măsură supus modelelor tipice jeleurilor. În special, unele modificări patologice ale organismelor vii sunt însoțite de fenomene de sinereză. Recent, s-a acordat multă atenție sistemelor apoase polimerice de tip jeleu (hidrogeluri), care sunt capabile să se umfle intens de zeci și sute de ori și să se prăbușească sub acțiunea electroliților, cu schimbări de temperatură și cu aplicarea câmpurilor electrice. Un exemplu de astfel de sisteme sunt jeleurile slab reticulate obținute pe bază de copolimeri de acid acrilic și acrilamidă. Ele sunt utilizate, în special, pentru a crea membrane cu permeabilitate controlată a substanțelor medicinale, ca absorbanți și, de asemenea, ca modele în analiza proceselor biologice. |
Substanțele chimice reale, cu care practic trebuie să se ocupe, și chiar cristalele ultrapure ale semiconductorilor elementari Ge și Si, conțin întotdeauna impurități reziduale, adică reprezintă întotdeauna substanțe formate din mai multe elemente chimice. Interacțiunea elementelor chimice care formează acest material poate fi foarte complexă. Rezultatul specific al acestei interacțiuni depinde de natura cristalo-chimică a elementelor care interacționează, de concentrația lor, precum și de factori externi - temperatură și presiune.
Principalele mijloace de reprezentare a rezultatelor interacțiunii elementelor chimice sau compușilor care formează o substanță dată sunt diagramele stării sistemului. Diagrama de stări prezintă stări stabile, adică stări care, în condiții date, au un minim de energie liberă. Prin urmare, o diagramă de stare poate fi numită și diagramă de echilibru de fază, deoarece arată care faze de echilibru există în condiții date. În conformitate cu aceasta, modificările stării sistemului, care sunt reflectate în diagramă, se referă la condiții de echilibru, adică în absența suprarăcirii sau suprasaturarii în sistem. Cu toate acestea, transformările de fază nu pot avea loc în condiții de echilibru (vezi mai jos), așa că diagrama de stare este un caz teoretic. Cu toate acestea, rolul diagramelor de stare în înțelegerea naturii și a rezultatelor interacțiunii diferitelor substanțe chimice și în prezicerea acestor rezultate este extrem de important, deoarece natura interacțiunii este cea care determină proprietățile materialului rezultat. În practică, diagramele de stare sunt utilizate pentru a lua în considerare transformările la viteze scăzute de răcire sau încălzire.
diagrama de stare sisteme se numește reprezentare geometrică a stărilor de fază de echilibru ale unui sistem termodinamic monocomponent sau multicomponent în funcție de parametrii care determină aceste stări (concentrație, temperatură, presiune).
Să definim câteva concepte folosite în descrierea diagramelor de stare.
sistem termodinamic numit corp de dimensiuni macroscopice (un set de corpuri), între ale căror părți individuale (între
între care) este posibil transferul de căldură şi difuzia a cel puţin uneia dintre componentele sistemului şi pentru care (care) sunt valabile legile termodinamicii.
Sistemele termodinamice sunt împărțite în omogenȘi eterogen. omogen se numește sistem termodinamic, în interiorul căruia nu există interfețe de fază care să separă părți ale sistemului unele de altele, care ar diferi fie prin structura cristalină, fie prin proprietățile lor fizice și chimice. eterogen sistemul constă din părți având fie o structură diferită, fie proprietăți fizico-chimice diferite și separate între ele prin interfețe de fază. Un exemplu de sistem eterogen este apa,
în echilibru cu aburul.
Fază- acesta este un sistem omogen sau un sistem care este o colecție de sisteme omogene identice ca structură cristalină și proprietăți fizico-chimice, separate între ele prin interfețe. În exemplul de mai sus, fazele sunt apă și abur, care diferă, de exemplu, prin densitate.
Interfețele de fază sunt straturi de grosime finită, în care cel puțin unul dintre parametrii sistemului se modifică în direcția de la o fază la alta. Interfețele fazelor în raport cu fazele adiacente au energie în exces (energie de tensiune superficială).
Pentru solide, cea mai importantă caracteristică a unei faze este rețeaua sa cristalină.1 Fiecare fază solidă are o rețea cristalină proprie, unică, care diferă de rețelele altor faze fie ca tip, fie ca parametri. Faza cristalină solidă poate fi obținută sub forma unui monocristal sau a unui policrist, care este o colecție de boabe sau cristalite. Cristalitele unui policristal, orientate diferit în spațiu, sunt separate între ele prin interfețe în mai multe straturi atomice (vezi cap. 3). Evident, granițele de cereale nu sunt limite de interfaz.
Sistemele termodinamice pot fi monocomponente sau multicomponente.
Componenta sistemului numită parte a sistemului, al cărei număr poate varia indiferent de numărul de alte părți. În cazul nostru, componentele sistemului pot fi elemente chimice sau compuși. Numărul componentelor sistemului, în general, poate să nu fie
1În principiu, faza solidă poate fi și amorfă sau sticloasă. Ambele faze sunt caracterizate prin absența ordinii pe distanță lungă în aranjamentul atomilor, asemănând mai degrabă cu un lichid. Aici vom lua în considerare doar materialele cristaline.

Orez. 4.1. Diagrama de stare a sistemului Ge–Si.
este egal cu numărul de elemente chimice diferite din sistem. De exemplu, apa (H2O) este formată din hidrogen și oxigen, dar este un sistem monocomponent. Pe fig. 4.1 și fig. 4.2 prezintă diagramele de echilibru de fază a două sisteme semiconductoare (binare) bicomponente caracteristice - Ge-Si și InSb-AlSb. Componentele sistemului în primul caz sunt Ge și Si, iar în al doilea caz, InSb și AlSb, și nu Sb, Al, In, deoarece cantitatea de In și Al din sistem depinde de cantitatea de Sb și cantitatea de InSb nu depinde de cantitatea de AlSb. De aceea numărul de componente ale sistemului este numărul minim de substanțe chimice necesare pentru a forma orice fază a unui sistem dat.
O stare de echilibru termodinamic a unui sistem este o stare în care parametrii acestei stări nu se modifică în timp și nu există fluxuri de orice tip în sistem.
Starea de echilibru a sistemului poate fi monofazată, bifazată și multifazată. Când două sau mai multe faze solide sunt amestecate, soluții solide, compuși și amestecuri mecanice. Acesta din urmă se realizează dacă aceste faze nu interacționează între ele. Fazele care formează un amestec pot fi elemente, compuși sau soluții solide pe bază de acestea, precum și modificări alotropice ale aceluiași element chimic (α și β-staniu etc.). Numărul maxim posibil de faze în echilibru este determinat de regula fazei Gibbs. Regula fazei stabilește relația dintre

Orez. 4.2. Diagrama de stare a sistemului InSb–AlSb.
prin numărul de faze, componente și grade de libertate ale sistemului:
c= k− f+ 2, (4.1)
Unde c- numărul de grade de libertate ale sistemului, k- numărul de componente ale sistemului, f- numărul de faze din sistem.
Sub numărul de grade de libertate sistemele înțeleg numărul de parametri externi și interni (temperatura, presiunea și concentrația), care pot fi modificați fără modificarea numărului de faze din sistem. Dacă numărul de grade de libertate este egal cu zero, atunci este imposibil să modificați parametrii externi și interni ai sistemului fără a provoca o modificare a numărului de faze. Dacă numărul de grade de libertate este egal cu unu, atunci este posibilă modificarea unuia dintre parametri în anumite limite și acest lucru nu va determina o scădere sau creștere a numărului de faze.
De exemplu, luați în considerare cazul cristalizării unei substanțe pure (un semiconductor elementar) la presiune constantă. În acest caz, regula Gibbs ia forma c= k− f+ 1.2 Când un semiconductor
este în stare lichidă f= 1, numărul de grade de libertate este 1 ( c= k− f+1 = 1 − 1 + 1 = 1). Temperatura în acest caz poate fi
schimbare fără a modifica starea de agregare. În momentul cristalizării
f= 2 (două faze - solid și lichid), c= k− f+1 = 1 − 2+1 = 0. Aceasta este
înseamnă că cele două faze sunt în echilibru la un strict definit
2 Variabilele independente din ecuația Gibbs sunt concentrația, temperatura și presiunea. Dacă presiunea este constantă, atunci numărul de variabile din ecuație va scădea cu una.
temperatura (punctul de topire) și nu poate fi modificată până când una dintre faze dispare (pe graficul temperatură-timp apare o zonă T= const, a cărui lungime va fi egală cu timpul de la începutul până la sfârșitul cristalizării). Sursa menținerii unei temperaturi constante în acest caz este eliberarea căldură latentă de cristalizare egală cu diferența dintre conținutul de căldură al fazelor vechi și noi. La terminarea cristalizării, în sistem rămâne doar o fază solidă, adică temperatura se poate schimba (scădea) din nou fără a modifica numărul de faze.
Diagramele de stare descriu compoziția de fază a sistemului la diferite concentrații de componente X, temperaturile T si presiune P. Diagramele de stare sunt în general spațiale. Dimensiunea spațiului depinde de numărul de variabile independente a căror funcție este compoziția de fază. Aceste variabile sunt coordonatele în care este construită diagrama. Cel mai simplu tip de diagramă de fază caracterizează starea unui material pur monocomponent în funcție de presiune și temperatură, de exemplu, binecunoscuta diagramă de stare a apei. Cu toate acestea, nu vom lua în considerare astfel de sisteme cu o singură componentă, ci vom trece imediat la luarea în considerare a sistemelor multicomponente, deoarece tocmai diagramele multicomponente sunt utilizate în producția de semiconductori. Cel mai adesea, astfel de diagrame sunt construite în coordonate temperatură-concentrație ( T− X). ÎN
În acest caz, pentru sistemele binare (cu două componente), diagramele sunt afișate pe un plan. Pentru sistemele ternare (cu trei componente), diagramele sunt construite în spațiu tridimensional etc. Dacă, pe lângă temperatură, presiunea este și ea variabilă, atunci chiar și pentru sistemele binare, diagramele devin tridimensionale ( P− T− X diagrame). În cele ce urmează, vom lua în considerare în principal numai sisteme binare construite în coordonate T− X. Totuși, acest capitol va discuta și P− T− X diagrame ale unor sisteme binare semiconductoare de mare importanţă practică.
De obicei, concentrația de pe diagrame este exprimată în greutate sau fracții molare ale unuia dintre componente sau în procente atomice. Prin urmare, aria de concentrare se schimbă reprezentată pe axă X, este limitată și se extinde de la zero la unu sau până la 100%. Pentru sistemele semiconductoare, împreună cu diagramele construite la scară liniară, se construiesc uneori diagrame în care concentrația unei componente este reprezentată în atomi pe centimetru cub sau în procente atomice, dar se folosește o scară logaritmică. Acest lucru se datorează faptului că, de regulă, solubilitatea limită (vezi cap. 7) este cea mai mare.

Orez. 4.3. Diagrama de stare a sistemului Si-Au cu diferite scale de-a lungul axei de concentrare (în regiunea adiacentă semiconductorului, procentul atomic al dopantului este reprezentat pe o scară logaritmică, iar apoi concentrația în procente atomice este reprezentată pe o scară liniară ).
Conținutul de elemente (impurități) în semiconductori în stare solidă este mic (mai puțin de 0,1 at.%) și dopajul efectiv utilizat în concentrație este de 1015–1019 atomi/cm3, adică 10−5–10−2 at. % (vezi Fig. 4.3).
Diagramele de fază ale stării oferă informații despre natura fazelor și compoziția de fază a sistemului cu o modificare a concentrației unuia sau mai multor componente, temperatură și presiune. Cu ajutorul diagramelor de stare de echilibru pentru condiții date, se pot determina: 1) numărul de faze din sistem; 2) compoziția fiecărei faze, natura acesteia (substanță elementară, compus, soluție solidă) și condițiile în care se formează; 3) cantitatea relativă a fiecăreia dintre faze.
Diagramele de fază sunt construite pe baza datelor de analiză fizică și chimică. Această analiză se bazează pe studiul experimental al dependențelor proprietăților fizice de parametri precum concentrația, temperatura și presiunea. Cunoașterea acestor dependențe permite stabilirea naturii fazelor și limitele existenței lor. Cele mai frecvente metode utilizate pentru construirea diagramelor de fază sunt metodele termografice și dilatometrice. Esența lor constă în faptul că, pentru un aliaj cu o compoziție dată, temperaturile transformărilor de fază sunt determinate de o modificare asemănătoare unui salt a entalpiei. H(conținut de căldură) sau volum V sistem, fixat pe curbele temperatură-timp (temperatura se notează la anumite intervale de timp) sau temperatură-volum în procesul de răcire sau încălzire a aliajului. După ce s-au determinat astfel punctele transformărilor de fază pentru aliajele de diferite compoziții ale unui sistem dat, este posibil să se construiască întreaga diagramă de fază. Aceste metode determină doar transformări de fază de primul fel. Aceste tranziții ar trebui să fie diferențiate de transformările de fază de al doilea fel (stări ferromagnetic-paramagnetice, supraconductoare-nesuperconductoare, ordonate-dezordonate), însoțite de o modificare asemănătoare unui salt a coeficientului de compresibilitate și a capacității termice. În acest caz, se construiesc diagrame compoziție-proprietate sau, pentru o compoziție dată, diagrame temperatură-proprietate etc.
Diagramele de echilibru de fază, sau diagramele de stare, într-o formă grafică convenabilă arată compoziția de fază a unui aliaj în funcție de temperatură și concentrație. Diagramele de stare sunt construite pentru condiții de echilibru sau condiții suficient de apropiate de acestea.
Starea de echilibru corespunde valorii minime a energiei Gibbs. Această stare poate fi atinsă numai la viteze de răcire foarte scăzute sau la încălzire prelungită. În acest sens, luarea în considerare a diagramelor de stare face posibilă determinarea transformărilor de fază în condiții de răcire sau încălzire foarte lentă. Echilibrul adevărat este rareori atins în condiții practice. În marea majoritate a cazurilor, aliajele sunt într-o stare metastabilă, adică într-o stare în care au stabilitate limitată și, sub influența factorilor externi, trec în alte stări mai stabile, deoarece energia lor Gibbs este mai mare decât minimul. În scopuri practice, este important ca stările metalabile să confere deseori proprietăți mecanice ridicate sau alte proprietăți aliajelor. În acest caz, știința metalelor trebuie să stabilească natura stărilor metastabile care oferă un set optim de proprietăți și să dezvolte moduri de tratament termic sau orice alt tratament care să facă posibilă obținerea acestor stări de neechilibru. Punctul de plecare în rezolvarea acestor probleme este cunoaşterea diagramelor de echilibru de fază.
regula fazei. Diagramele de echilibru de fază caracterizează starea finală sau limită a aliajelor, adică, obținută după ce toate transformările lor au avut loc și s-au încheiat complet. Această stare a aliajului depinde de condițiile externe (temperatură, presiune) și se caracterizează prin numărul și concentrația fazelor formate. Modelul de schimbare a numărului de faze într-un sistem eterogen este determinat de regula fazelor.
Regula fazelor stabilește relația dintre numărul de grade de libertate, numărul de componente și numărul de faze și este exprimată prin ecuație
unde C este numărul de grade de libertate ale sistemului (sau varianță); K - numărul de componente care formează sistemul, adică numărul minim de elemente chimice necesare formării oricărei faze a sistemului; 2 - numărul de factori externi; Ф este numărul de faze aflate în echilibru.
Numarul de grade de libertate (varianta sistemului) se intelege ca fiind posibilitatea modificarii temperaturii, presiunii si concentratiei fara modificarea numarului de faze aflate in echilibru.
Când se studiază echilibrele fizice și chimice, temperatura și presiunea sunt considerate factori externi care afectează starea aliajului. Aplicând regula fazei metalelor, în multe cazuri este posibil să acceptăm ca schimbare doar un singur factor extern - temperatura, deoarece presiunea, cu excepția presiunii foarte mari, are un efect redus asupra echilibrului de fază al aliajelor în stare solidă și lichidă. Atunci ecuația va lua următoarea formă: . Deoarece numărul de grade de libertate nu poate fi mai mic decât zero și nu poate fi un număr fracțional, atunci, adică, numărul de faze dintr-un aliaj aflat în echilibru nu poate fi mai mare decât numărul de componente plus unu. În consecință, nu mai mult de trei faze pot fi în echilibru într-un sistem binar, nu mai mult de patru într-un sistem ternar etc.
Dacă numărul maxim de faze este în echilibru într-un sistem cu un anumit număr de componente, atunci numărul de grade de libertate al sistemului este egal cu zero.Un astfel de echilibru se numește invariant (nonvariant). La echilibru invariant, un aliaj dintr-un anumit număr de faze nu poate exista decât în condiții foarte specifice: la o temperatură constantă și o anumită compoziție a tuturor fazelor aflate în echilibru. Aceasta înseamnă că transformarea începe și se termină la aceeași temperatură constantă.
În cazul unei scăderi a numărului de faze cu una față de numărul maxim posibil de grade de libertate crește cu unu.Un astfel de sistem se numește monovariant (o-variantă). Când sistemul este bivariant (două variante).
Echilibrul în sistemele cu două componente. După cum sa menționat deja, condiția de echilibru este minim liber

Orez. 34. Dependența energiei Gibbs (energia liberă) de compoziția aliajului
energie. Doar acele procese fizice apar spontan în sistem, în care energia liberă scade. Dacă aliajul constă dintr-o singură fază, de exemplu, o soluție lichidă sau solidă a, atunci energia Gibbs la temperatură și presiune constante depinde de natura sa și de compoziția fazei (Fig. 34, a). Pentru cazul prezentat în fig. 34, a, soluția solidă a este stabilă, deoarece energia sa Gibbs este mai mică decât cea a fazei lichide
Dacă un sistem (aliaj) este format din două sau mai multe faze, atunci la temperatură și presiune constante, energia lui Gibbs este determinată de regula de amestecare (Fig. 34, b).
Punctul (Fig. 34, b) care caracterizează energia Gibbs a aliajului de compoziție se află pe o linie dreaptă care leagă punctele care caracterizează energia Gibbs a fazelor a și - și împarte această linie dreaptă în segmente invers proporționale cu cantitățile de masă ale - faze.
Dacă fazele a- și - care formează acest sistem își pot schimba compoziția, atunci energia Gibbs a fiecărei faze, în funcție de concentrație, se poate modifica așa cum se arată în Fig. 34, c.
Compoziția fazelor aflate în echilibru la o temperatură dată corespunde punctelor (Fig. 34, c). Starea în două faze corespunde concentrațiilor care se află în interiorul cărora energia Gibbs a unui amestec de două faze - compoziție și - compoziție determinată de puncte de pe o linie dreaptă este mai mică decât energia liberă a fazelor individuale. Compozițiile cu o concentrație mai mică decât în condiții de echilibru vor consta numai din faza - și aliaje cu o concentrație mai mare - din faza -.
Într-un sistem cu două componente, în anumite condiții, de exemplu, la echilibru invariant, trei faze pot coexista simultan, de exemplu, o fază lichidă și două soluții solide.
Izotermele de energie liberă în funcție de compoziție pentru acest caz sunt prezentate în Fig. 34, d. Compoziția fazelor aflate în echilibru este determinată de proiecția pe axa concentrației a punctelor de contact ale dreptei - la curbe (puncte
Analiza termică este utilizată pentru a construi diagrame de fază, în special pentru a determina temperaturile de solidificare. În acest scop, curbele de răcire ale aliajelor individuale sunt obținute experimental, iar temperaturile transformărilor corespunzătoare sunt determinate din inflexiunile sau opririle acestora asociate cu efectele termice ale transformărilor. Aceste temperaturi sunt numite puncte critice. Pentru studiul transformărilor în stare solidă se folosesc diverse metode de analiză fizico-chimică: microanaliză, difracție de raze X, dilatometrică, magnetică etc.